{"title":"survivveai:基于体细胞RNA-Seq表达的癌症患者长期生存预测。","authors":"Omri Nayshool, Nitzan Kol, Elisheva Javaski, Ninette Amariglio, Gideon Rechavi","doi":"10.1177/11769351221127875","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Prediction of cancer outcome is a major challenge in oncology and is essential for treatment planning. Repositories such as The Cancer Genome Atlas (TCGA) contain vast amounts of data for many types of cancers. Our goal was to create reliable prediction models using TCGA data and validate them using an external dataset.</p><p><strong>Results: </strong>For 16 TCGA cancer type cohorts we have optimized a Random Forest prediction model using parameter grid search followed by a backward feature elimination loop for dimensions reduction. For each feature that was removed, the model was retrained and the area under the curve of the receiver operating characteristic (AUC-ROC) was calculated using test data. Five prediction models gave AUC-ROC bigger than 80%. We used Clinical Proteomic Tumor Analysis Consortium v3 (CPTAC3) data for validation. The most enriched pathways for the top models were those involved in basic functions related to tumorigenesis and organ development. Enrichment for 2 prediction models of the TCGA-KIRP cohort was explored, one with 42 genes (AUC-ROC = 0.86) the other is composed of 300 genes (AUC-ROC = 0.85). The most enriched networks for both models share only 5 network nodes: DMBT1, IL11, HOXB6, TRIB3, PIM1. These genes play a significant role in renal cancer and might be used for prognosis prediction and as candidate therapeutic targets.</p><p><strong>Availability and implementation: </strong>The prediction models were created and tested using Python SciKit-Learn package. They are freely accessible via a friendly web interface we called surviveAI at https://tinyurl.com/surviveai.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":" ","pages":"11769351221127875"},"PeriodicalIF":2.5000,"publicationDate":"2022-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7a/c4/10.1177_11769351221127875.PMC9549197.pdf","citationCount":"1","resultStr":"{\"title\":\"SurviveAI: Long Term Survival Prediction of Cancer Patients Based on Somatic RNA-Seq Expression.\",\"authors\":\"Omri Nayshool, Nitzan Kol, Elisheva Javaski, Ninette Amariglio, Gideon Rechavi\",\"doi\":\"10.1177/11769351221127875\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Prediction of cancer outcome is a major challenge in oncology and is essential for treatment planning. Repositories such as The Cancer Genome Atlas (TCGA) contain vast amounts of data for many types of cancers. Our goal was to create reliable prediction models using TCGA data and validate them using an external dataset.</p><p><strong>Results: </strong>For 16 TCGA cancer type cohorts we have optimized a Random Forest prediction model using parameter grid search followed by a backward feature elimination loop for dimensions reduction. For each feature that was removed, the model was retrained and the area under the curve of the receiver operating characteristic (AUC-ROC) was calculated using test data. Five prediction models gave AUC-ROC bigger than 80%. We used Clinical Proteomic Tumor Analysis Consortium v3 (CPTAC3) data for validation. The most enriched pathways for the top models were those involved in basic functions related to tumorigenesis and organ development. Enrichment for 2 prediction models of the TCGA-KIRP cohort was explored, one with 42 genes (AUC-ROC = 0.86) the other is composed of 300 genes (AUC-ROC = 0.85). The most enriched networks for both models share only 5 network nodes: DMBT1, IL11, HOXB6, TRIB3, PIM1. These genes play a significant role in renal cancer and might be used for prognosis prediction and as candidate therapeutic targets.</p><p><strong>Availability and implementation: </strong>The prediction models were created and tested using Python SciKit-Learn package. They are freely accessible via a friendly web interface we called surviveAI at https://tinyurl.com/surviveai.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\" \",\"pages\":\"11769351221127875\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2022-10-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7a/c4/10.1177_11769351221127875.PMC9549197.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351221127875\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351221127875","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
SurviveAI: Long Term Survival Prediction of Cancer Patients Based on Somatic RNA-Seq Expression.
Motivation: Prediction of cancer outcome is a major challenge in oncology and is essential for treatment planning. Repositories such as The Cancer Genome Atlas (TCGA) contain vast amounts of data for many types of cancers. Our goal was to create reliable prediction models using TCGA data and validate them using an external dataset.
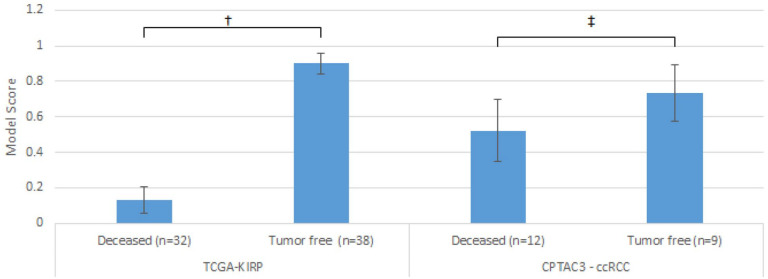
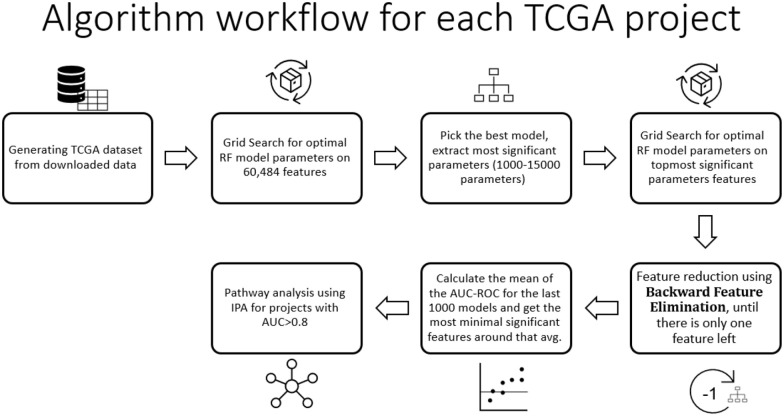
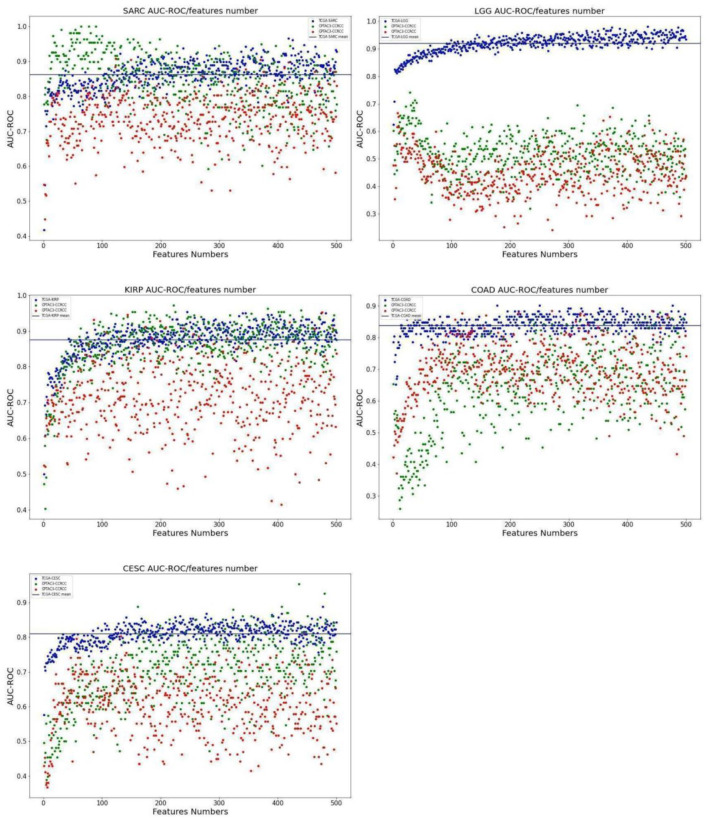
Results: For 16 TCGA cancer type cohorts we have optimized a Random Forest prediction model using parameter grid search followed by a backward feature elimination loop for dimensions reduction. For each feature that was removed, the model was retrained and the area under the curve of the receiver operating characteristic (AUC-ROC) was calculated using test data. Five prediction models gave AUC-ROC bigger than 80%. We used Clinical Proteomic Tumor Analysis Consortium v3 (CPTAC3) data for validation. The most enriched pathways for the top models were those involved in basic functions related to tumorigenesis and organ development. Enrichment for 2 prediction models of the TCGA-KIRP cohort was explored, one with 42 genes (AUC-ROC = 0.86) the other is composed of 300 genes (AUC-ROC = 0.85). The most enriched networks for both models share only 5 network nodes: DMBT1, IL11, HOXB6, TRIB3, PIM1. These genes play a significant role in renal cancer and might be used for prognosis prediction and as candidate therapeutic targets.
Availability and implementation: The prediction models were created and tested using Python SciKit-Learn package. They are freely accessible via a friendly web interface we called surviveAI at https://tinyurl.com/surviveai.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


