Wafaa Bouzroud, Amal Tazzite, Sarah Berrada, Bouchaïb Gazzaz, Hind Dehbi
{"title":"MECP2基因R306X突变导致摩洛哥患者非典型Rett综合征:一例报告","authors":"Wafaa Bouzroud, Amal Tazzite, Sarah Berrada, Bouchaïb Gazzaz, Hind Dehbi","doi":"10.1177/2632010X221124269","DOIUrl":null,"url":null,"abstract":"<p><p>Rett syndrome (RTT) is a rare X-linked syndrome that predominantly affects girls. It is characterized by a severe and progressive neurodevelopmental disorder with neurological regression and autism spectrum features. The Rett syndrome is associated with a broad phenotypic spectrum. It ranges from a classical Rett syndrome defined by well-established criteria to atypical cases with symptoms similar to other syndromes, such as Angelman syndrome. The first case of a Moroccan female child carrying a R306X mutation in the <i>MECP2</i> (Methyl-CpG-Binding Protein 2) gene, with an unusual manifestation of Rett syndrome, is presented here. She showed autistic regression, behavioral stagnation, epilepsy, unmotivated laughter, and craniofacial dysmorphia. Whole exome sequencing revealed a nonsense mutation (R306X), resulting in a truncated, nonfunctional MECP2 protein. The overlapping phenotypic spectrums between Rett and Angelman syndromes have been described, and an interaction between the <i>MECP2</i> gene and the <i>UBE3A</i> (Ubiquitin Protein Ligase E3A) gene pathways is possible but has not yet been proven. An extensive genetic analysis is highly recommended in atypical cases to ensure an accurate diagnosis and to improve patient management and genetic counseling.</p>","PeriodicalId":53204,"journal":{"name":"Clinical Pathology","volume":" ","pages":"2632010X221124269"},"PeriodicalIF":1.9000,"publicationDate":"2022-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/48/f9/10.1177_2632010X221124269.PMC9486266.pdf","citationCount":"0","resultStr":"{\"title\":\"R306X Mutation in the <i>MECP2</i> Gene Causes an Atypical Rett Syndrome in a Moroccan Patient: A Case Report.\",\"authors\":\"Wafaa Bouzroud, Amal Tazzite, Sarah Berrada, Bouchaïb Gazzaz, Hind Dehbi\",\"doi\":\"10.1177/2632010X221124269\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Rett syndrome (RTT) is a rare X-linked syndrome that predominantly affects girls. It is characterized by a severe and progressive neurodevelopmental disorder with neurological regression and autism spectrum features. The Rett syndrome is associated with a broad phenotypic spectrum. It ranges from a classical Rett syndrome defined by well-established criteria to atypical cases with symptoms similar to other syndromes, such as Angelman syndrome. The first case of a Moroccan female child carrying a R306X mutation in the <i>MECP2</i> (Methyl-CpG-Binding Protein 2) gene, with an unusual manifestation of Rett syndrome, is presented here. She showed autistic regression, behavioral stagnation, epilepsy, unmotivated laughter, and craniofacial dysmorphia. Whole exome sequencing revealed a nonsense mutation (R306X), resulting in a truncated, nonfunctional MECP2 protein. The overlapping phenotypic spectrums between Rett and Angelman syndromes have been described, and an interaction between the <i>MECP2</i> gene and the <i>UBE3A</i> (Ubiquitin Protein Ligase E3A) gene pathways is possible but has not yet been proven. An extensive genetic analysis is highly recommended in atypical cases to ensure an accurate diagnosis and to improve patient management and genetic counseling.</p>\",\"PeriodicalId\":53204,\"journal\":{\"name\":\"Clinical Pathology\",\"volume\":\" \",\"pages\":\"2632010X221124269\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2022-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/48/f9/10.1177_2632010X221124269.PMC9486266.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pathology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/2632010X221124269\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"PATHOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pathology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/2632010X221124269","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"PATHOLOGY","Score":null,"Total":0}
R306X Mutation in the MECP2 Gene Causes an Atypical Rett Syndrome in a Moroccan Patient: A Case Report.
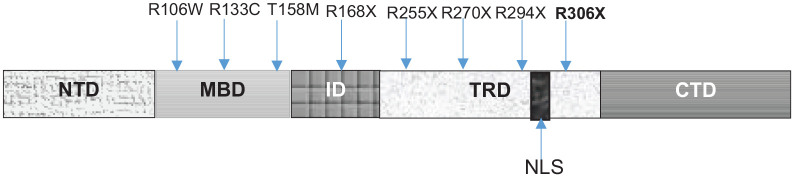
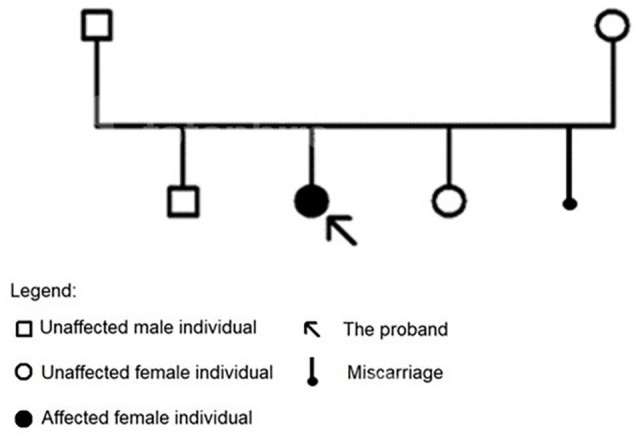
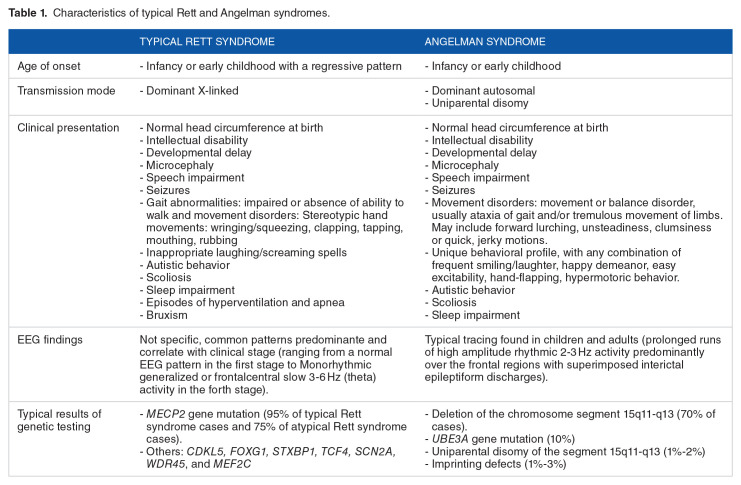
Rett syndrome (RTT) is a rare X-linked syndrome that predominantly affects girls. It is characterized by a severe and progressive neurodevelopmental disorder with neurological regression and autism spectrum features. The Rett syndrome is associated with a broad phenotypic spectrum. It ranges from a classical Rett syndrome defined by well-established criteria to atypical cases with symptoms similar to other syndromes, such as Angelman syndrome. The first case of a Moroccan female child carrying a R306X mutation in the MECP2 (Methyl-CpG-Binding Protein 2) gene, with an unusual manifestation of Rett syndrome, is presented here. She showed autistic regression, behavioral stagnation, epilepsy, unmotivated laughter, and craniofacial dysmorphia. Whole exome sequencing revealed a nonsense mutation (R306X), resulting in a truncated, nonfunctional MECP2 protein. The overlapping phenotypic spectrums between Rett and Angelman syndromes have been described, and an interaction between the MECP2 gene and the UBE3A (Ubiquitin Protein Ligase E3A) gene pathways is possible but has not yet been proven. An extensive genetic analysis is highly recommended in atypical cases to ensure an accurate diagnosis and to improve patient management and genetic counseling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


