Jiafeng Yao, Hao Gu, Wenjun Mou, Zhenping Chen, Jie Ma, Honghao Ma, Nan Li, Rui Zhang, Tianyou Wang, Jin Jiang, Runhui Wu
{"title":"复合杂合变异LRBA基因的多种表型:儿童细胞减少症病例系列报道。","authors":"Jiafeng Yao, Hao Gu, Wenjun Mou, Zhenping Chen, Jie Ma, Honghao Ma, Nan Li, Rui Zhang, Tianyou Wang, Jin Jiang, Runhui Wu","doi":"10.1177/03946320221125591","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>LPS-responsive beige-like anchor (LRBA) deficiency is one of the most common monogenic disorders causing common variable immunodeficiency (CVID) and CVID-like disorders. However, the clinical spectrum of compound heterozygous (CHZ) LRBA variation should be extended. In this study, we presented five cases of compound heterozygous LRBA with various refractory cytopenias.</p><p><strong>Materials and methods: </strong>Retrospective analysis of the clinical manifestations, management, and outcomes of five cases (from five pedigrees) with <i>LRBA</i> gene CHZ variants which initially manifested as single/multilineage immune cytopenias was performed.</p><p><strong>Results: </strong>1. Gene variations: All five patients inherited the compound heterozygous LRBA variations from their parents which were thought to be pathogenic. BEACH, DUF4704, and LamG were the main affected domains of LRBA gene in this case series. 2. Immune dysregulation of clinic: (1) Hypogammaglobulinemia were recorded in four patients, and the proportion of Treg was decreased in two patients. Only one patient had been with increased TCRαβ+CD4/CD8 double-negative T cells (DNT). (2) Lymphoproliferative manifestations were seen in three patients. (3) All five patients were complained with cytopenia, although they showed different clinical manifestations. None of the parents was asymptomatic. (4) Other immune disorders: P5 also had relapsed infections and autoimmune endocrinopathy. 3. Management and outcomes: P1 and P5 responded well to immunomodulatory therapy and P3 was effectively treated with hemophagocytic lymphohistiocytosis (HLH) first-line regimen chemotherapy. P4 showed no responses to steroids and IVIG. However, TPO-R agonist was effective.</p><p><strong>Conclusion: </strong>Unlike homozygous mutations, compound heterozygous LRBA variation should always be kept in mind for the various phenotypes and different treatment responses.</p>","PeriodicalId":14046,"journal":{"name":"International Journal of Immunopathology and Pharmacology","volume":"36 ","pages":"3946320221125591"},"PeriodicalIF":2.6000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/4f/fb/10.1177_03946320221125591.PMC9465590.pdf","citationCount":"1","resultStr":"{\"title\":\"Various phenotypes of <i>LRBA</i> gene with compound heterozygous variation: A case series report of pediatric cytopenia patients.\",\"authors\":\"Jiafeng Yao, Hao Gu, Wenjun Mou, Zhenping Chen, Jie Ma, Honghao Ma, Nan Li, Rui Zhang, Tianyou Wang, Jin Jiang, Runhui Wu\",\"doi\":\"10.1177/03946320221125591\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>LPS-responsive beige-like anchor (LRBA) deficiency is one of the most common monogenic disorders causing common variable immunodeficiency (CVID) and CVID-like disorders. However, the clinical spectrum of compound heterozygous (CHZ) LRBA variation should be extended. In this study, we presented five cases of compound heterozygous LRBA with various refractory cytopenias.</p><p><strong>Materials and methods: </strong>Retrospective analysis of the clinical manifestations, management, and outcomes of five cases (from five pedigrees) with <i>LRBA</i> gene CHZ variants which initially manifested as single/multilineage immune cytopenias was performed.</p><p><strong>Results: </strong>1. Gene variations: All five patients inherited the compound heterozygous LRBA variations from their parents which were thought to be pathogenic. BEACH, DUF4704, and LamG were the main affected domains of LRBA gene in this case series. 2. Immune dysregulation of clinic: (1) Hypogammaglobulinemia were recorded in four patients, and the proportion of Treg was decreased in two patients. Only one patient had been with increased TCRαβ+CD4/CD8 double-negative T cells (DNT). (2) Lymphoproliferative manifestations were seen in three patients. (3) All five patients were complained with cytopenia, although they showed different clinical manifestations. None of the parents was asymptomatic. (4) Other immune disorders: P5 also had relapsed infections and autoimmune endocrinopathy. 3. Management and outcomes: P1 and P5 responded well to immunomodulatory therapy and P3 was effectively treated with hemophagocytic lymphohistiocytosis (HLH) first-line regimen chemotherapy. P4 showed no responses to steroids and IVIG. However, TPO-R agonist was effective.</p><p><strong>Conclusion: </strong>Unlike homozygous mutations, compound heterozygous LRBA variation should always be kept in mind for the various phenotypes and different treatment responses.</p>\",\"PeriodicalId\":14046,\"journal\":{\"name\":\"International Journal of Immunopathology and Pharmacology\",\"volume\":\"36 \",\"pages\":\"3946320221125591\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/4f/fb/10.1177_03946320221125591.PMC9465590.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Immunopathology and Pharmacology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1177/03946320221125591\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Immunopathology and Pharmacology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1177/03946320221125591","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Various phenotypes of LRBA gene with compound heterozygous variation: A case series report of pediatric cytopenia patients.
Objective: LPS-responsive beige-like anchor (LRBA) deficiency is one of the most common monogenic disorders causing common variable immunodeficiency (CVID) and CVID-like disorders. However, the clinical spectrum of compound heterozygous (CHZ) LRBA variation should be extended. In this study, we presented five cases of compound heterozygous LRBA with various refractory cytopenias.
Materials and methods: Retrospective analysis of the clinical manifestations, management, and outcomes of five cases (from five pedigrees) with LRBA gene CHZ variants which initially manifested as single/multilineage immune cytopenias was performed.
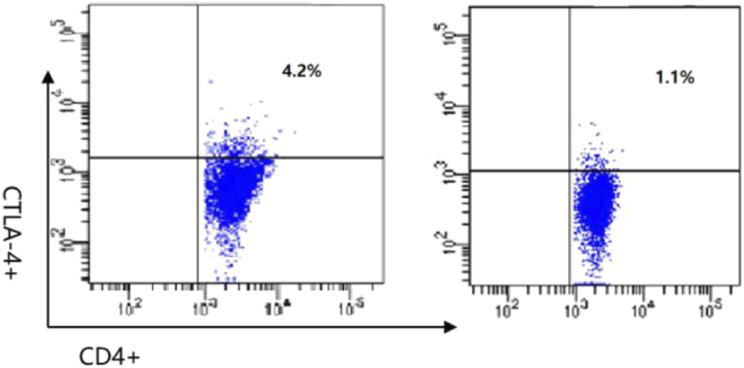
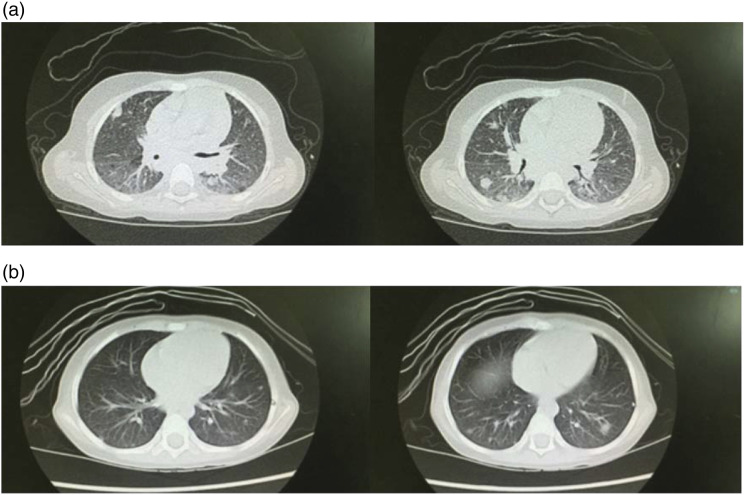
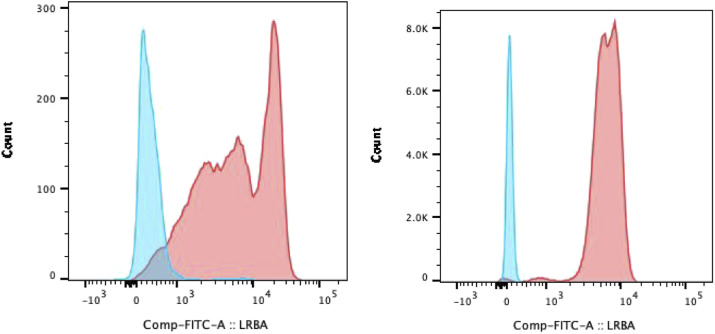
Results: 1. Gene variations: All five patients inherited the compound heterozygous LRBA variations from their parents which were thought to be pathogenic. BEACH, DUF4704, and LamG were the main affected domains of LRBA gene in this case series. 2. Immune dysregulation of clinic: (1) Hypogammaglobulinemia were recorded in four patients, and the proportion of Treg was decreased in two patients. Only one patient had been with increased TCRαβ+CD4/CD8 double-negative T cells (DNT). (2) Lymphoproliferative manifestations were seen in three patients. (3) All five patients were complained with cytopenia, although they showed different clinical manifestations. None of the parents was asymptomatic. (4) Other immune disorders: P5 also had relapsed infections and autoimmune endocrinopathy. 3. Management and outcomes: P1 and P5 responded well to immunomodulatory therapy and P3 was effectively treated with hemophagocytic lymphohistiocytosis (HLH) first-line regimen chemotherapy. P4 showed no responses to steroids and IVIG. However, TPO-R agonist was effective.
Conclusion: Unlike homozygous mutations, compound heterozygous LRBA variation should always be kept in mind for the various phenotypes and different treatment responses.
期刊介绍:
International Journal of Immunopathology and Pharmacology is an Open Access peer-reviewed journal publishing original papers describing research in the fields of immunology, pathology and pharmacology. The intention is that the journal should reflect both the experimental and clinical aspects of immunology as well as advances in the understanding of the pathology and pharmacology of the immune system.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


