J R Deller, Hayder Radha, J Justin McCormick, Huiyan Wang
{"title":"发现差异表达基因的非线性依赖。","authors":"J R Deller, Hayder Radha, J Justin McCormick, Huiyan Wang","doi":"10.5402/2012/564715","DOIUrl":null,"url":null,"abstract":"<p><p>Microarray data are used to determine which genes are active in response to a changing cell environment. Genes are \"discovered\" when they are significantly differentially expressed in the microarray data collected under the differing conditions. In one prevalent approach, all genes are assumed to satisfy a null hypothesis, ℍ 0, of no difference in expression. A false discovery (type 1 error) occurs when ℍ 0 is incorrectly rejected. The quality of a detection algorithm is assessed by estimating its number of false discoveries, 𝔉. Work involving the second-moment modeling of the z-value histogram (representing gene expression differentials) has shown significantly deleterious effects of intergene expression correlation on the estimate of 𝔉. This paper suggests that nonlinear dependencies could likewise be important. With an applied emphasis, this paper extends the \"moment framework\" by including third-moment skewness corrections in an estimator of 𝔉. This estimator combines observed correlation (corrected for sampling fluctuations) with the information from easily identifiable null cases. Nonlinear-dependence modeling reduces the estimation error relative to that of linear estimation. Third-moment calculations involve empirical densities of 3 × 3 covariance matrices estimated using very few samples. The principle of entropy maximization is employed to connect estimated moments to 𝔉 inference. Model results are tested with BRCA and HIV data sets and with carefully constructed simulations. </p>","PeriodicalId":90877,"journal":{"name":"ISRN bioinformatics","volume":"2012 ","pages":"564715"},"PeriodicalIF":0.0000,"publicationDate":"2012-04-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4393074/pdf/","citationCount":"2","resultStr":"{\"title\":\"Nonlinear dependence in the discovery of differentially expressed genes.\",\"authors\":\"J R Deller, Hayder Radha, J Justin McCormick, Huiyan Wang\",\"doi\":\"10.5402/2012/564715\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Microarray data are used to determine which genes are active in response to a changing cell environment. Genes are \\\"discovered\\\" when they are significantly differentially expressed in the microarray data collected under the differing conditions. In one prevalent approach, all genes are assumed to satisfy a null hypothesis, ℍ 0, of no difference in expression. A false discovery (type 1 error) occurs when ℍ 0 is incorrectly rejected. The quality of a detection algorithm is assessed by estimating its number of false discoveries, 𝔉. Work involving the second-moment modeling of the z-value histogram (representing gene expression differentials) has shown significantly deleterious effects of intergene expression correlation on the estimate of 𝔉. This paper suggests that nonlinear dependencies could likewise be important. With an applied emphasis, this paper extends the \\\"moment framework\\\" by including third-moment skewness corrections in an estimator of 𝔉. This estimator combines observed correlation (corrected for sampling fluctuations) with the information from easily identifiable null cases. Nonlinear-dependence modeling reduces the estimation error relative to that of linear estimation. Third-moment calculations involve empirical densities of 3 × 3 covariance matrices estimated using very few samples. The principle of entropy maximization is employed to connect estimated moments to 𝔉 inference. Model results are tested with BRCA and HIV data sets and with carefully constructed simulations. </p>\",\"PeriodicalId\":90877,\"journal\":{\"name\":\"ISRN bioinformatics\",\"volume\":\"2012 \",\"pages\":\"564715\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-04-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4393074/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ISRN bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5402/2012/564715\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2012/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5402/2012/564715","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2012/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Nonlinear dependence in the discovery of differentially expressed genes.
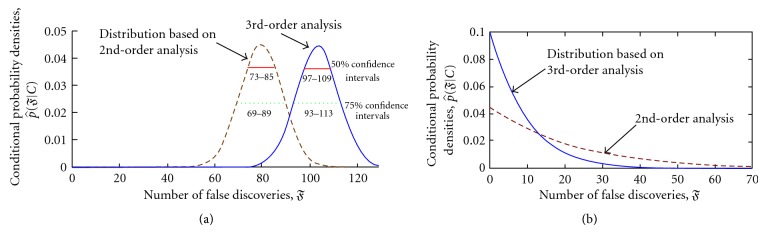
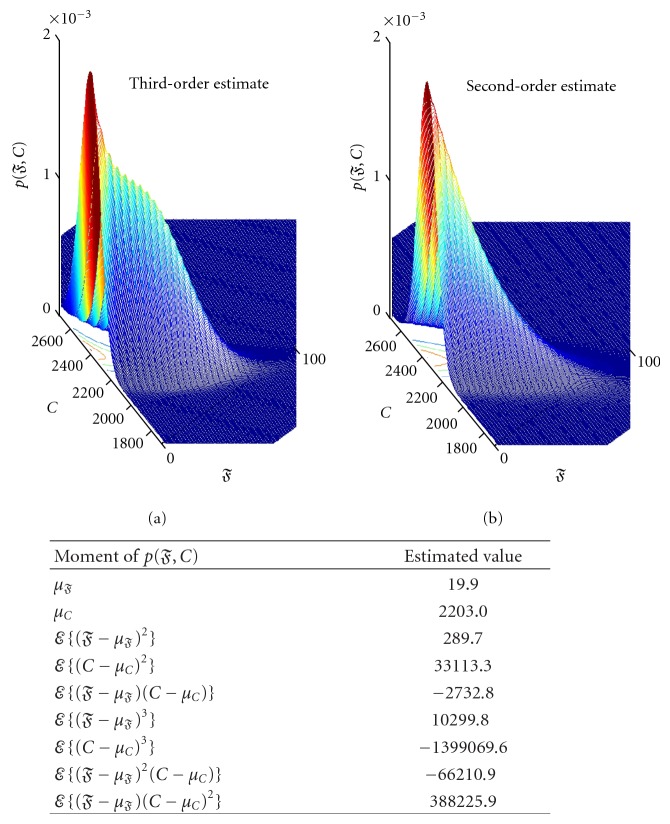
Microarray data are used to determine which genes are active in response to a changing cell environment. Genes are "discovered" when they are significantly differentially expressed in the microarray data collected under the differing conditions. In one prevalent approach, all genes are assumed to satisfy a null hypothesis, ℍ 0, of no difference in expression. A false discovery (type 1 error) occurs when ℍ 0 is incorrectly rejected. The quality of a detection algorithm is assessed by estimating its number of false discoveries, 𝔉. Work involving the second-moment modeling of the z-value histogram (representing gene expression differentials) has shown significantly deleterious effects of intergene expression correlation on the estimate of 𝔉. This paper suggests that nonlinear dependencies could likewise be important. With an applied emphasis, this paper extends the "moment framework" by including third-moment skewness corrections in an estimator of 𝔉. This estimator combines observed correlation (corrected for sampling fluctuations) with the information from easily identifiable null cases. Nonlinear-dependence modeling reduces the estimation error relative to that of linear estimation. Third-moment calculations involve empirical densities of 3 × 3 covariance matrices estimated using very few samples. The principle of entropy maximization is employed to connect estimated moments to 𝔉 inference. Model results are tested with BRCA and HIV data sets and with carefully constructed simulations.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


