Saumya K Patel, Linz-Buoy George, Sivakumar Prasanth Kumar, Hyacinth N Highland, Yogesh T Jasrai, Himanshu A Pandya, Ketaki R Desai
{"title":"恶性疟原虫多药耐药蛋白研究的计算方法","authors":"Saumya K Patel, Linz-Buoy George, Sivakumar Prasanth Kumar, Hyacinth N Highland, Yogesh T Jasrai, Himanshu A Pandya, Ketaki R Desai","doi":"10.1155/2013/437168","DOIUrl":null,"url":null,"abstract":"<p><p>The emergence of drug resistance in Plasmodium falciparum tremendously affected the chemotherapy worldwide while the intense distribution of chloroquine-resistant strains in most of the endemic areas added more complications in the treatment of malaria. The situation has even worsened by the lack of molecular mechanism to understand the resistance conferred by Plasmodia species. Recent studies have suggested the association of antimalarial resistance with P. falciparum multidrug resistance protein 1 (PfMDR1), an ATP-binding cassette (ABC) transporter and a homologue of human P-glycoprotein 1 (P-gp1). The present study deals about the development of PfMDR1 computational model and the model of substrate transport across PfMDR1 with insights derived from conformations relative to inward- and outward-facing topologies that switch on/off the transportation system. Comparison of ATP docked positions and its structural motif binding properties were found to be similar among other ATPases, and thereby contributes to NBD domains dimerization, a unique structural agreement noticed in Mus musculus Pgp and Escherichia coli MDR transporter homolog (MsbA). The interaction of leading antimalarials and phytochemicals within the active pocket of both wild-type and mutant-type PfMDR1 demonstrated the mode of binding and provided insights of less binding affinity thereby contributing to parasite's resistance mechanism. </p>","PeriodicalId":90877,"journal":{"name":"ISRN bioinformatics","volume":"2013 ","pages":"437168"},"PeriodicalIF":0.0000,"publicationDate":"2013-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4393060/pdf/","citationCount":"6","resultStr":"{\"title\":\"A Computational Approach towards the Understanding of Plasmodium falciparum Multidrug Resistance Protein 1.\",\"authors\":\"Saumya K Patel, Linz-Buoy George, Sivakumar Prasanth Kumar, Hyacinth N Highland, Yogesh T Jasrai, Himanshu A Pandya, Ketaki R Desai\",\"doi\":\"10.1155/2013/437168\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The emergence of drug resistance in Plasmodium falciparum tremendously affected the chemotherapy worldwide while the intense distribution of chloroquine-resistant strains in most of the endemic areas added more complications in the treatment of malaria. The situation has even worsened by the lack of molecular mechanism to understand the resistance conferred by Plasmodia species. Recent studies have suggested the association of antimalarial resistance with P. falciparum multidrug resistance protein 1 (PfMDR1), an ATP-binding cassette (ABC) transporter and a homologue of human P-glycoprotein 1 (P-gp1). The present study deals about the development of PfMDR1 computational model and the model of substrate transport across PfMDR1 with insights derived from conformations relative to inward- and outward-facing topologies that switch on/off the transportation system. Comparison of ATP docked positions and its structural motif binding properties were found to be similar among other ATPases, and thereby contributes to NBD domains dimerization, a unique structural agreement noticed in Mus musculus Pgp and Escherichia coli MDR transporter homolog (MsbA). The interaction of leading antimalarials and phytochemicals within the active pocket of both wild-type and mutant-type PfMDR1 demonstrated the mode of binding and provided insights of less binding affinity thereby contributing to parasite's resistance mechanism. </p>\",\"PeriodicalId\":90877,\"journal\":{\"name\":\"ISRN bioinformatics\",\"volume\":\"2013 \",\"pages\":\"437168\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4393060/pdf/\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ISRN bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2013/437168\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2013/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/437168","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
A Computational Approach towards the Understanding of Plasmodium falciparum Multidrug Resistance Protein 1.
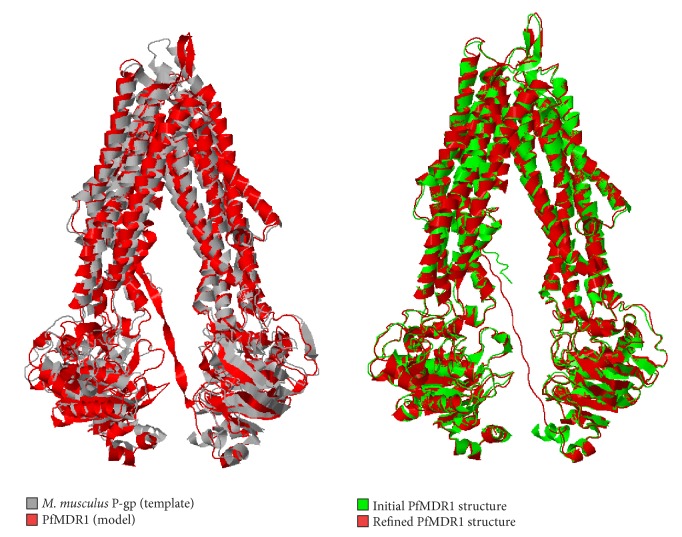
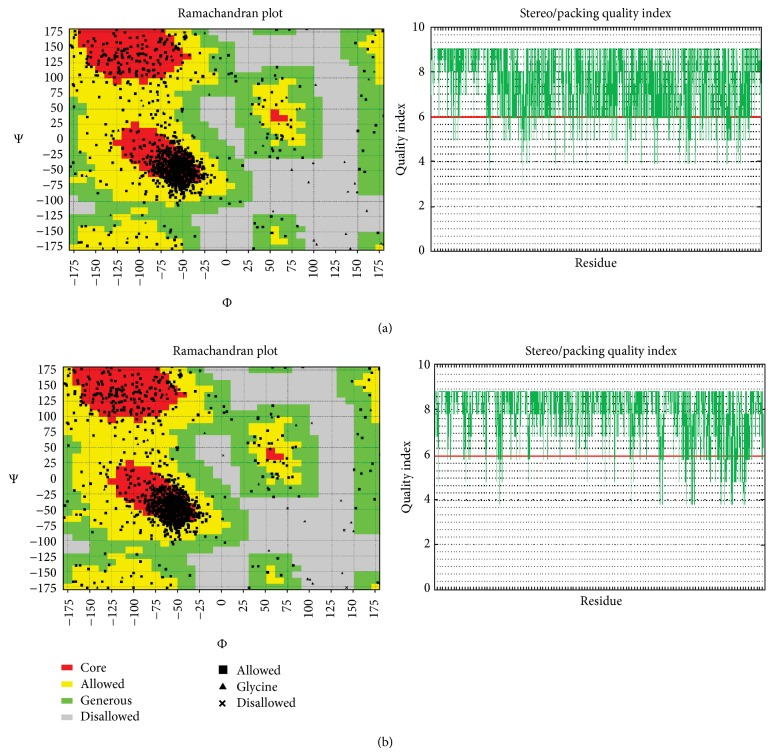
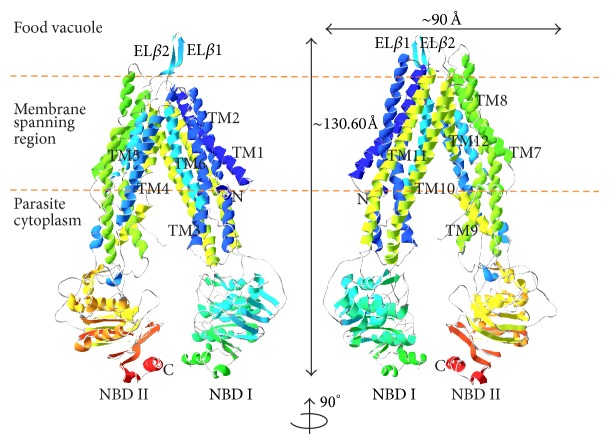
The emergence of drug resistance in Plasmodium falciparum tremendously affected the chemotherapy worldwide while the intense distribution of chloroquine-resistant strains in most of the endemic areas added more complications in the treatment of malaria. The situation has even worsened by the lack of molecular mechanism to understand the resistance conferred by Plasmodia species. Recent studies have suggested the association of antimalarial resistance with P. falciparum multidrug resistance protein 1 (PfMDR1), an ATP-binding cassette (ABC) transporter and a homologue of human P-glycoprotein 1 (P-gp1). The present study deals about the development of PfMDR1 computational model and the model of substrate transport across PfMDR1 with insights derived from conformations relative to inward- and outward-facing topologies that switch on/off the transportation system. Comparison of ATP docked positions and its structural motif binding properties were found to be similar among other ATPases, and thereby contributes to NBD domains dimerization, a unique structural agreement noticed in Mus musculus Pgp and Escherichia coli MDR transporter homolog (MsbA). The interaction of leading antimalarials and phytochemicals within the active pocket of both wild-type and mutant-type PfMDR1 demonstrated the mode of binding and provided insights of less binding affinity thereby contributing to parasite's resistance mechanism.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


