{"title":"利用可识别性和基因间相关性来改进差异表达的检测。","authors":"J R Deller, Hayder Radha, J Justin McCormick","doi":"10.1155/2013/404717","DOIUrl":null,"url":null,"abstract":"<p><p>Accurate differential analysis of microarray data strongly depends on effective treatment of intergene correlation. Such dependence is ordinarily accounted for in terms of its effect on significance cutoffs. In this paper, it is shown that correlation can, in fact, be exploited to share information across tests and reorder expression differentials for increased statistical power, regardless of the threshold. Significantly improved differential analysis is the result of two simple measures: (i) adjusting test statistics to exploit information from identifiable genes (the large subset of genes represented on a microarray that can be classified a priori as nondifferential with very high confidence], but (ii) doing so in a way that accounts for linear dependencies among identifiable and nonidentifiable genes. A method is developed that builds upon the widely used two-sample t-statistic approach and uses analysis in Hilbert space to decompose the nonidentified gene vector into two components that are correlated and uncorrelated with the identified set. In the application to data derived from a widely studied prostate cancer database, the proposed method outperforms some of the most highly regarded approaches published to date. Algorithms in MATLAB and in R are available for public download. </p>","PeriodicalId":90877,"journal":{"name":"ISRN bioinformatics","volume":"2013 ","pages":"404717"},"PeriodicalIF":0.0000,"publicationDate":"2013-06-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2013/404717","citationCount":"0","resultStr":"{\"title\":\"Exploiting identifiability and intergene correlation for improved detection of differential expression.\",\"authors\":\"J R Deller, Hayder Radha, J Justin McCormick\",\"doi\":\"10.1155/2013/404717\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Accurate differential analysis of microarray data strongly depends on effective treatment of intergene correlation. Such dependence is ordinarily accounted for in terms of its effect on significance cutoffs. In this paper, it is shown that correlation can, in fact, be exploited to share information across tests and reorder expression differentials for increased statistical power, regardless of the threshold. Significantly improved differential analysis is the result of two simple measures: (i) adjusting test statistics to exploit information from identifiable genes (the large subset of genes represented on a microarray that can be classified a priori as nondifferential with very high confidence], but (ii) doing so in a way that accounts for linear dependencies among identifiable and nonidentifiable genes. A method is developed that builds upon the widely used two-sample t-statistic approach and uses analysis in Hilbert space to decompose the nonidentified gene vector into two components that are correlated and uncorrelated with the identified set. In the application to data derived from a widely studied prostate cancer database, the proposed method outperforms some of the most highly regarded approaches published to date. Algorithms in MATLAB and in R are available for public download. </p>\",\"PeriodicalId\":90877,\"journal\":{\"name\":\"ISRN bioinformatics\",\"volume\":\"2013 \",\"pages\":\"404717\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-06-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2013/404717\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ISRN bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2013/404717\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2013/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/404717","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Exploiting identifiability and intergene correlation for improved detection of differential expression.
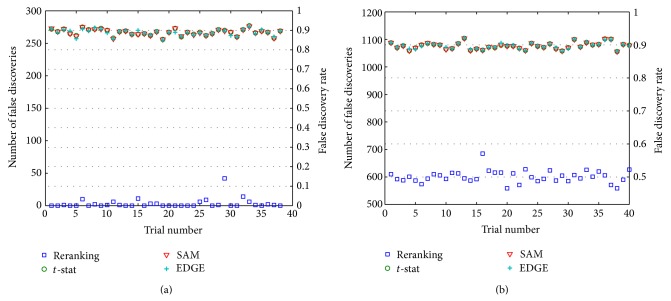
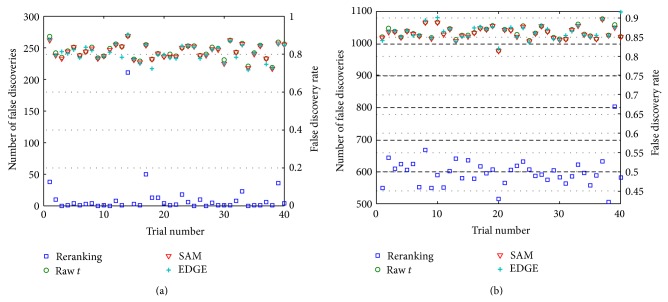
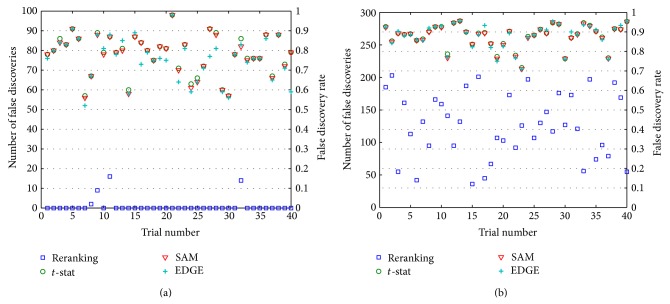
Accurate differential analysis of microarray data strongly depends on effective treatment of intergene correlation. Such dependence is ordinarily accounted for in terms of its effect on significance cutoffs. In this paper, it is shown that correlation can, in fact, be exploited to share information across tests and reorder expression differentials for increased statistical power, regardless of the threshold. Significantly improved differential analysis is the result of two simple measures: (i) adjusting test statistics to exploit information from identifiable genes (the large subset of genes represented on a microarray that can be classified a priori as nondifferential with very high confidence], but (ii) doing so in a way that accounts for linear dependencies among identifiable and nonidentifiable genes. A method is developed that builds upon the widely used two-sample t-statistic approach and uses analysis in Hilbert space to decompose the nonidentified gene vector into two components that are correlated and uncorrelated with the identified set. In the application to data derived from a widely studied prostate cancer database, the proposed method outperforms some of the most highly regarded approaches published to date. Algorithms in MATLAB and in R are available for public download.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


