{"title":"hSOD1拷贝数的减少显著影响G37R(29号线)小鼠的ALS表型表现:对推定治疗剂评估的影响。","authors":"Pierre Zwiegers, Grace Lee, Christopher A Shaw","doi":"10.1186/1477-5751-13-14","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>In vivo animal models of familial amyotrophic lateral sclerosis (fALS) are widely used to delineate the potential role that genetic mutations play in the neurodegenerative process. While these models are extensively used for establishing the safety and efficacy of putative therapeutics during pre-clinical development, effective clinical translation of pharmacological interventions has been largely unsuccessful.</p><p><strong>Results: </strong>In this report we compare a recent cohort of G37R (line 29) mice generated from mating wild-type females with transgenic males obtained commercially to a previous set of offspring produced with transgenic male breeders from a colony established at a local collaborator's facility. Commercially derived progeny presented with a tightly clustered genomic signature for the mutant human superoxide dismutase1 transgene (hSOD1) locus, and exhibited a greater than two-fold reduction in the number of transgene copies present in the genome compared to offspring derived locally. Decrease in transgene levels corresponded with delayed ALS progression and a significant increase in overall lifespan (146%).</p><p><strong>Conclusions: </strong>These results highlight some key challenges inherent to the use of G37R (line 29) animals in pre-clinical studies for the development of ALS therapeutics. Without stringent assessment of mutant SOD1 copy number/protein levels, heterogeneity of transgene levels within cohorts may influence the behavioural and pathological presentation of disease and thus calls to question the validity of any detected therapeutic effects. Nuanced changes in mutant SOD1 copy number that currently remain unreported may undermine research endeavours, delay efforts for clinical translation, and compromise the rigor of animal studies by limiting reproducibility amongst research groups.</p>","PeriodicalId":73849,"journal":{"name":"Journal of negative results in biomedicine","volume":"13 ","pages":"14"},"PeriodicalIF":0.0000,"publicationDate":"2014-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1477-5751-13-14","citationCount":"16","resultStr":"{\"title\":\"Reduction in hSOD1 copy number significantly impacts ALS phenotype presentation in G37R (line 29) mice: implications for the assessment of putative therapeutic agents.\",\"authors\":\"Pierre Zwiegers, Grace Lee, Christopher A Shaw\",\"doi\":\"10.1186/1477-5751-13-14\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>In vivo animal models of familial amyotrophic lateral sclerosis (fALS) are widely used to delineate the potential role that genetic mutations play in the neurodegenerative process. While these models are extensively used for establishing the safety and efficacy of putative therapeutics during pre-clinical development, effective clinical translation of pharmacological interventions has been largely unsuccessful.</p><p><strong>Results: </strong>In this report we compare a recent cohort of G37R (line 29) mice generated from mating wild-type females with transgenic males obtained commercially to a previous set of offspring produced with transgenic male breeders from a colony established at a local collaborator's facility. Commercially derived progeny presented with a tightly clustered genomic signature for the mutant human superoxide dismutase1 transgene (hSOD1) locus, and exhibited a greater than two-fold reduction in the number of transgene copies present in the genome compared to offspring derived locally. Decrease in transgene levels corresponded with delayed ALS progression and a significant increase in overall lifespan (146%).</p><p><strong>Conclusions: </strong>These results highlight some key challenges inherent to the use of G37R (line 29) animals in pre-clinical studies for the development of ALS therapeutics. Without stringent assessment of mutant SOD1 copy number/protein levels, heterogeneity of transgene levels within cohorts may influence the behavioural and pathological presentation of disease and thus calls to question the validity of any detected therapeutic effects. Nuanced changes in mutant SOD1 copy number that currently remain unreported may undermine research endeavours, delay efforts for clinical translation, and compromise the rigor of animal studies by limiting reproducibility amongst research groups.</p>\",\"PeriodicalId\":73849,\"journal\":{\"name\":\"Journal of negative results in biomedicine\",\"volume\":\"13 \",\"pages\":\"14\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2014-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1477-5751-13-14\",\"citationCount\":\"16\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of negative results in biomedicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1477-5751-13-14\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of negative results in biomedicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1477-5751-13-14","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Reduction in hSOD1 copy number significantly impacts ALS phenotype presentation in G37R (line 29) mice: implications for the assessment of putative therapeutic agents.
Background: In vivo animal models of familial amyotrophic lateral sclerosis (fALS) are widely used to delineate the potential role that genetic mutations play in the neurodegenerative process. While these models are extensively used for establishing the safety and efficacy of putative therapeutics during pre-clinical development, effective clinical translation of pharmacological interventions has been largely unsuccessful.
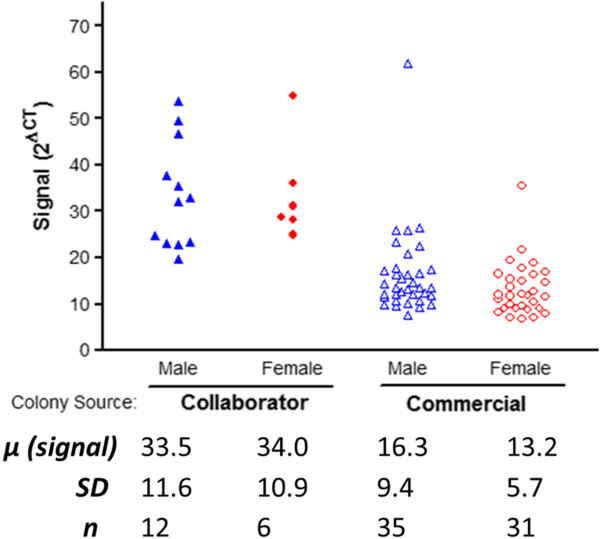
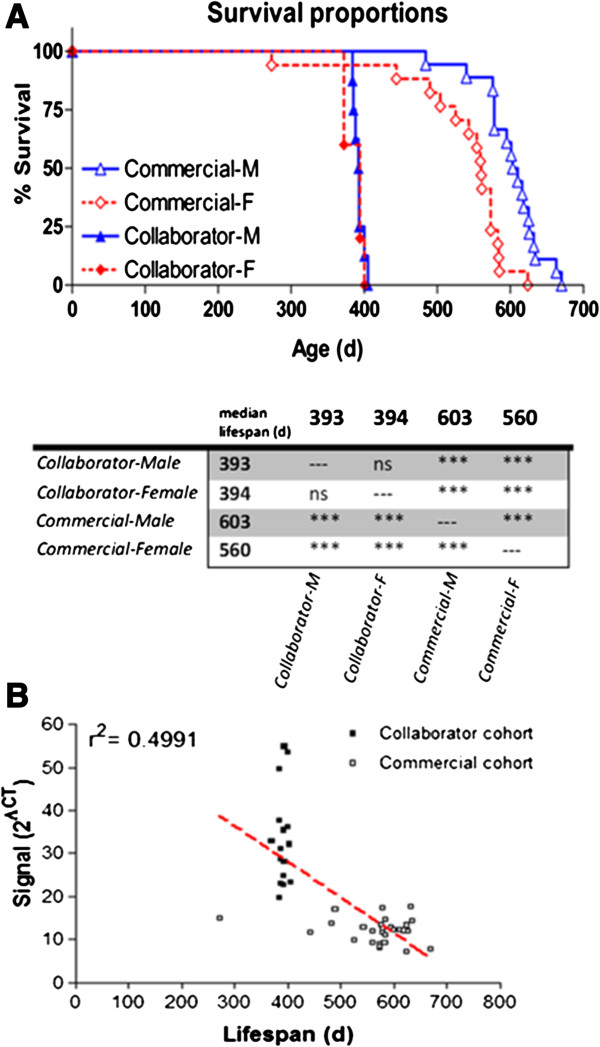
Results: In this report we compare a recent cohort of G37R (line 29) mice generated from mating wild-type females with transgenic males obtained commercially to a previous set of offspring produced with transgenic male breeders from a colony established at a local collaborator's facility. Commercially derived progeny presented with a tightly clustered genomic signature for the mutant human superoxide dismutase1 transgene (hSOD1) locus, and exhibited a greater than two-fold reduction in the number of transgene copies present in the genome compared to offspring derived locally. Decrease in transgene levels corresponded with delayed ALS progression and a significant increase in overall lifespan (146%).
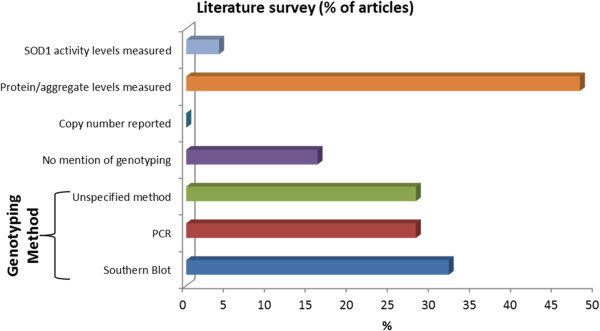
Conclusions: These results highlight some key challenges inherent to the use of G37R (line 29) animals in pre-clinical studies for the development of ALS therapeutics. Without stringent assessment of mutant SOD1 copy number/protein levels, heterogeneity of transgene levels within cohorts may influence the behavioural and pathological presentation of disease and thus calls to question the validity of any detected therapeutic effects. Nuanced changes in mutant SOD1 copy number that currently remain unreported may undermine research endeavours, delay efforts for clinical translation, and compromise the rigor of animal studies by limiting reproducibility amongst research groups.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


