Neda Zamani, Görel Sundström, Marc P Höppner, Manfred G Grabherr
{"title":"模块化和可配置的最佳序列校准软件:可乐。","authors":"Neda Zamani, Görel Sundström, Marc P Höppner, Manfred G Grabherr","doi":"10.1186/1751-0473-9-12","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The fundamental challenge in optimally aligning homologous sequences is to define a scoring scheme that best reflects the underlying biological processes. Maximising the overall number of matches in the alignment does not always reflect the patterns by which nucleotides mutate. Efficiently implemented algorithms that can be parameterised to accommodate more complex non-linear scoring schemes are thus desirable.</p><p><strong>Results: </strong>We present Cola, alignment software that implements different optimal alignment algorithms, also allowing for scoring contiguous matches of nucleotides in a nonlinear manner. The latter places more emphasis on short, highly conserved motifs, and less on the surrounding nucleotides, which can be more diverged. To illustrate the differences, we report results from aligning 14,100 sequences from 3' untranslated regions of human genes to 25 of their mammalian counterparts, where we found that a nonlinear scoring scheme is more consistent than a linear scheme in detecting short, conserved motifs.</p><p><strong>Conclusions: </strong>Cola is freely available under LPGL from https://github.com/nedaz/cola.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":"9 ","pages":"12"},"PeriodicalIF":0.0000,"publicationDate":"2014-06-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1751-0473-9-12","citationCount":"2","resultStr":"{\"title\":\"Modular and configurable optimal sequence alignment software: Cola.\",\"authors\":\"Neda Zamani, Görel Sundström, Marc P Höppner, Manfred G Grabherr\",\"doi\":\"10.1186/1751-0473-9-12\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The fundamental challenge in optimally aligning homologous sequences is to define a scoring scheme that best reflects the underlying biological processes. Maximising the overall number of matches in the alignment does not always reflect the patterns by which nucleotides mutate. Efficiently implemented algorithms that can be parameterised to accommodate more complex non-linear scoring schemes are thus desirable.</p><p><strong>Results: </strong>We present Cola, alignment software that implements different optimal alignment algorithms, also allowing for scoring contiguous matches of nucleotides in a nonlinear manner. The latter places more emphasis on short, highly conserved motifs, and less on the surrounding nucleotides, which can be more diverged. To illustrate the differences, we report results from aligning 14,100 sequences from 3' untranslated regions of human genes to 25 of their mammalian counterparts, where we found that a nonlinear scoring scheme is more consistent than a linear scheme in detecting short, conserved motifs.</p><p><strong>Conclusions: </strong>Cola is freely available under LPGL from https://github.com/nedaz/cola.</p>\",\"PeriodicalId\":35052,\"journal\":{\"name\":\"Source Code for Biology and Medicine\",\"volume\":\"9 \",\"pages\":\"12\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2014-06-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1751-0473-9-12\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Source Code for Biology and Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1751-0473-9-12\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2014/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Decision Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1751-0473-9-12","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2014/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
Modular and configurable optimal sequence alignment software: Cola.
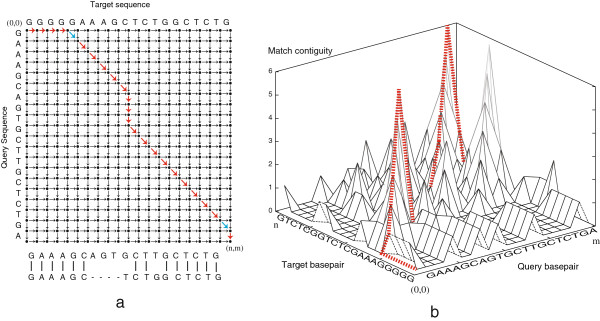
Background: The fundamental challenge in optimally aligning homologous sequences is to define a scoring scheme that best reflects the underlying biological processes. Maximising the overall number of matches in the alignment does not always reflect the patterns by which nucleotides mutate. Efficiently implemented algorithms that can be parameterised to accommodate more complex non-linear scoring schemes are thus desirable.
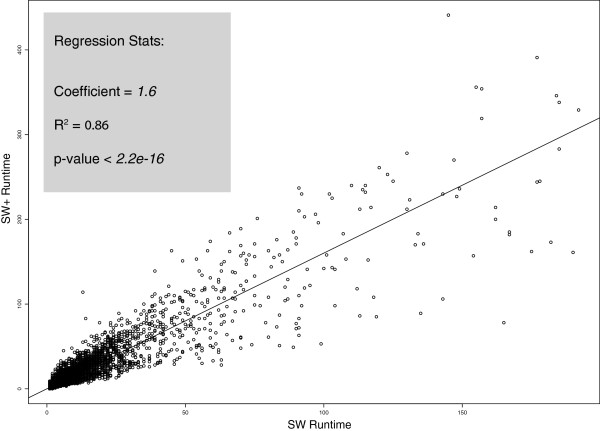
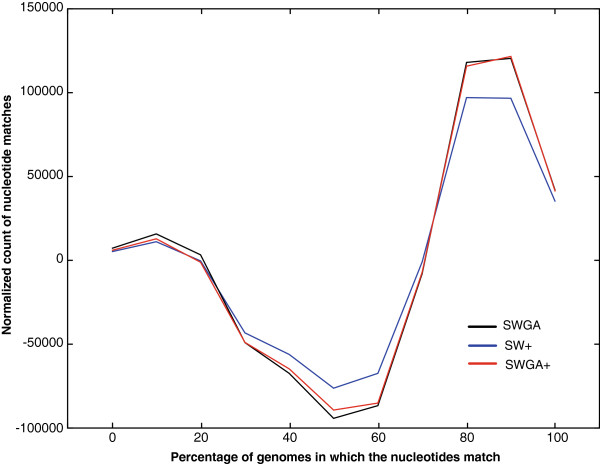
Results: We present Cola, alignment software that implements different optimal alignment algorithms, also allowing for scoring contiguous matches of nucleotides in a nonlinear manner. The latter places more emphasis on short, highly conserved motifs, and less on the surrounding nucleotides, which can be more diverged. To illustrate the differences, we report results from aligning 14,100 sequences from 3' untranslated regions of human genes to 25 of their mammalian counterparts, where we found that a nonlinear scoring scheme is more consistent than a linear scheme in detecting short, conserved motifs.
Conclusions: Cola is freely available under LPGL from https://github.com/nedaz/cola.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


