Mohammadmersad Ghorbani, Simon J E Taylor, Mark A Pook, Annette Payne
{"title":"三核苷酸重复扩增病DNA甲基化状态的比较(计算)分析。","authors":"Mohammadmersad Ghorbani, Simon J E Taylor, Mark A Pook, Annette Payne","doi":"10.1155/2013/689798","DOIUrl":null,"url":null,"abstract":"<p><p>Previous studies have examined DNA methylation in different trinucleotide repeat diseases. We have combined this data and used a pattern searching algorithm to identify motifs in the DNA surrounding aberrantly methylated CpGs found in the DNA of patients with one of the three trinucleotide repeat (TNR) expansion diseases: fragile X syndrome (FRAXA), myotonic dystrophy type I (DM1), or Friedreich's ataxia (FRDA). We examined sequences surrounding both the variably methylated (VM) CpGs, which are hypermethylated in patients compared with unaffected controls, and the nonvariably methylated CpGs which remain either always methylated (AM) or never methylated (NM) in both patients and controls. Using the J48 algorithm of WEKA analysis, we identified that two patterns are all that is necessary to classify our three regions CCGG∗ which is found in VM and not in AM regions and AATT∗ which distinguished between NM and VM + AM using proportional frequency. Furthermore, comparing our software with MEME software, we have demonstrated that our software identifies more patterns than MEME in these short DNA sequences. Thus, we present evidence that the DNA sequence surrounding CpG can influence its susceptibility to be de novo methylated in a disease state associated with a trinucleotide repeat. </p>","PeriodicalId":16575,"journal":{"name":"Journal of Nucleic Acids","volume":"2013 ","pages":"689798"},"PeriodicalIF":1.3000,"publicationDate":"2013-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2013/689798","citationCount":"15","resultStr":"{\"title\":\"Comparative (computational) analysis of the DNA methylation status of trinucleotide repeat expansion diseases.\",\"authors\":\"Mohammadmersad Ghorbani, Simon J E Taylor, Mark A Pook, Annette Payne\",\"doi\":\"10.1155/2013/689798\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Previous studies have examined DNA methylation in different trinucleotide repeat diseases. We have combined this data and used a pattern searching algorithm to identify motifs in the DNA surrounding aberrantly methylated CpGs found in the DNA of patients with one of the three trinucleotide repeat (TNR) expansion diseases: fragile X syndrome (FRAXA), myotonic dystrophy type I (DM1), or Friedreich's ataxia (FRDA). We examined sequences surrounding both the variably methylated (VM) CpGs, which are hypermethylated in patients compared with unaffected controls, and the nonvariably methylated CpGs which remain either always methylated (AM) or never methylated (NM) in both patients and controls. Using the J48 algorithm of WEKA analysis, we identified that two patterns are all that is necessary to classify our three regions CCGG∗ which is found in VM and not in AM regions and AATT∗ which distinguished between NM and VM + AM using proportional frequency. Furthermore, comparing our software with MEME software, we have demonstrated that our software identifies more patterns than MEME in these short DNA sequences. Thus, we present evidence that the DNA sequence surrounding CpG can influence its susceptibility to be de novo methylated in a disease state associated with a trinucleotide repeat. </p>\",\"PeriodicalId\":16575,\"journal\":{\"name\":\"Journal of Nucleic Acids\",\"volume\":\"2013 \",\"pages\":\"689798\"},\"PeriodicalIF\":1.3000,\"publicationDate\":\"2013-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2013/689798\",\"citationCount\":\"15\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Nucleic Acids\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2013/689798\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2013/12/23 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Nucleic Acids","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/689798","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/12/23 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Comparative (computational) analysis of the DNA methylation status of trinucleotide repeat expansion diseases.
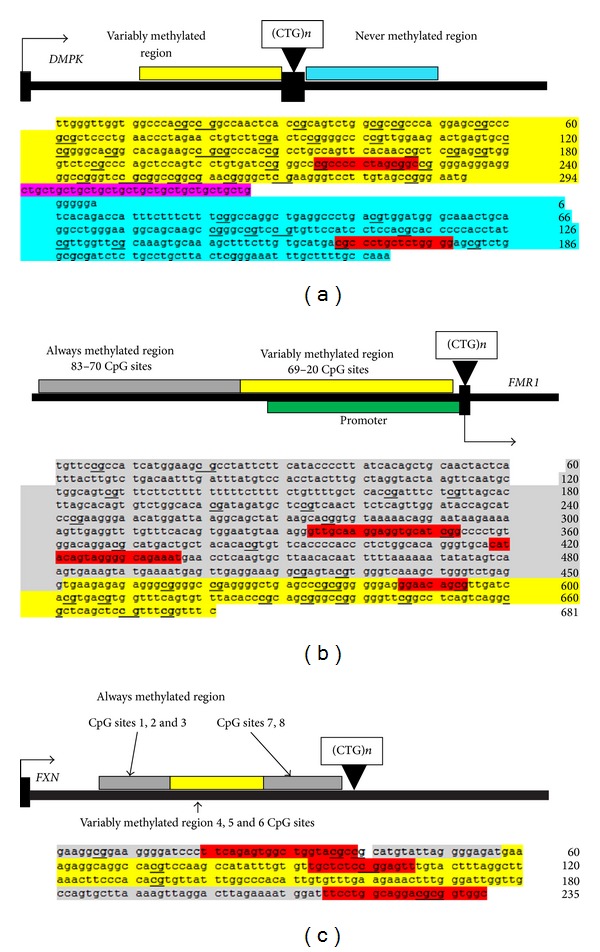
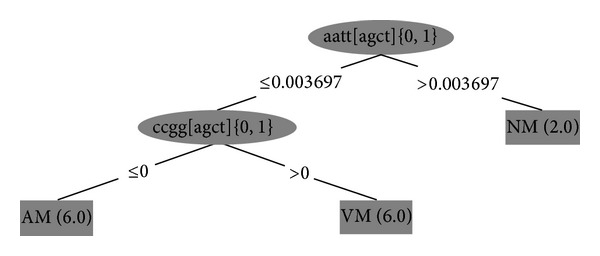
Previous studies have examined DNA methylation in different trinucleotide repeat diseases. We have combined this data and used a pattern searching algorithm to identify motifs in the DNA surrounding aberrantly methylated CpGs found in the DNA of patients with one of the three trinucleotide repeat (TNR) expansion diseases: fragile X syndrome (FRAXA), myotonic dystrophy type I (DM1), or Friedreich's ataxia (FRDA). We examined sequences surrounding both the variably methylated (VM) CpGs, which are hypermethylated in patients compared with unaffected controls, and the nonvariably methylated CpGs which remain either always methylated (AM) or never methylated (NM) in both patients and controls. Using the J48 algorithm of WEKA analysis, we identified that two patterns are all that is necessary to classify our three regions CCGG∗ which is found in VM and not in AM regions and AATT∗ which distinguished between NM and VM + AM using proportional frequency. Furthermore, comparing our software with MEME software, we have demonstrated that our software identifies more patterns than MEME in these short DNA sequences. Thus, we present evidence that the DNA sequence surrounding CpG can influence its susceptibility to be de novo methylated in a disease state associated with a trinucleotide repeat.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


