Chenwei Wang, Alperen Taciroglu, Stefan R Maetschke, Colleen C Nelson, Mark A Ragan, Melissa J Davis
{"title":"mCOPA:分析肿瘤表达数据的异质性特征。","authors":"Chenwei Wang, Alperen Taciroglu, Stefan R Maetschke, Colleen C Nelson, Mark A Ragan, Melissa J Davis","doi":"10.1186/2043-9113-2-22","DOIUrl":null,"url":null,"abstract":"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>Cancer outlier profile analysis (COPA) has proven to be an effective approach to analyzing cancer expression data, leading to the discovery of the TMPRSS2 and ETS family gene fusion events in prostate cancer. However, the original COPA algorithm did not identify down-regulated outliers, and the currently available R package implementing the method is similarly restricted to the analysis of over-expressed outliers. Here we present a modified outlier detection method, mCOPA, which contains refinements to the outlier-detection algorithm, identifies both over- and under-expressed outliers, is freely available, and can be applied to any expression dataset.</p><p><strong>Results: </strong>We compare our method to other feature-selection approaches, and demonstrate that mCOPA frequently selects more-informative features than do differential expression or variance-based feature selection approaches, and is able to recover observed clinical subtypes more consistently. We demonstrate the application of mCOPA to prostate cancer expression data, and explore the use of outliers in clustering, pathway analysis, and the identification of tumour suppressors. We analyse the under-expressed outliers to identify known and novel prostate cancer tumour suppressor genes, validating these against data in Oncomine and the Cancer Gene Index. We also demonstrate how a combination of outlier analysis and pathway analysis can identify molecular mechanisms disrupted in individual tumours.</p><p><strong>Conclusions: </strong>We demonstrate that mCOPA offers advantages, compared to differential expression or variance, in selecting outlier features, and that the features so selected are better able to assign samples to clinically annotated subtypes. Further, we show that the biology explored by outlier analysis differs from that uncovered in differential expression or variance analysis. mCOPA is an important new tool for the exploration of cancer datasets and the discovery of new cancer subtypes, and can be combined with pathway and functional analysis approaches to discover mechanisms underpinning heterogeneity in cancers.</p>","PeriodicalId":73663,"journal":{"name":"Journal of clinical bioinformatics","volume":"2 1","pages":"22"},"PeriodicalIF":0.0000,"publicationDate":"2012-12-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2043-9113-2-22","citationCount":"17","resultStr":"{\"title\":\"mCOPA: analysis of heterogeneous features in cancer expression data.\",\"authors\":\"Chenwei Wang, Alperen Taciroglu, Stefan R Maetschke, Colleen C Nelson, Mark A Ragan, Melissa J Davis\",\"doi\":\"10.1186/2043-9113-2-22\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>Cancer outlier profile analysis (COPA) has proven to be an effective approach to analyzing cancer expression data, leading to the discovery of the TMPRSS2 and ETS family gene fusion events in prostate cancer. However, the original COPA algorithm did not identify down-regulated outliers, and the currently available R package implementing the method is similarly restricted to the analysis of over-expressed outliers. Here we present a modified outlier detection method, mCOPA, which contains refinements to the outlier-detection algorithm, identifies both over- and under-expressed outliers, is freely available, and can be applied to any expression dataset.</p><p><strong>Results: </strong>We compare our method to other feature-selection approaches, and demonstrate that mCOPA frequently selects more-informative features than do differential expression or variance-based feature selection approaches, and is able to recover observed clinical subtypes more consistently. We demonstrate the application of mCOPA to prostate cancer expression data, and explore the use of outliers in clustering, pathway analysis, and the identification of tumour suppressors. We analyse the under-expressed outliers to identify known and novel prostate cancer tumour suppressor genes, validating these against data in Oncomine and the Cancer Gene Index. We also demonstrate how a combination of outlier analysis and pathway analysis can identify molecular mechanisms disrupted in individual tumours.</p><p><strong>Conclusions: </strong>We demonstrate that mCOPA offers advantages, compared to differential expression or variance, in selecting outlier features, and that the features so selected are better able to assign samples to clinically annotated subtypes. Further, we show that the biology explored by outlier analysis differs from that uncovered in differential expression or variance analysis. mCOPA is an important new tool for the exploration of cancer datasets and the discovery of new cancer subtypes, and can be combined with pathway and functional analysis approaches to discover mechanisms underpinning heterogeneity in cancers.</p>\",\"PeriodicalId\":73663,\"journal\":{\"name\":\"Journal of clinical bioinformatics\",\"volume\":\"2 1\",\"pages\":\"22\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-12-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2043-9113-2-22\",\"citationCount\":\"17\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of clinical bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2043-9113-2-22\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2043-9113-2-22","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
mCOPA: analysis of heterogeneous features in cancer expression data.
Unlabelled:
Background: Cancer outlier profile analysis (COPA) has proven to be an effective approach to analyzing cancer expression data, leading to the discovery of the TMPRSS2 and ETS family gene fusion events in prostate cancer. However, the original COPA algorithm did not identify down-regulated outliers, and the currently available R package implementing the method is similarly restricted to the analysis of over-expressed outliers. Here we present a modified outlier detection method, mCOPA, which contains refinements to the outlier-detection algorithm, identifies both over- and under-expressed outliers, is freely available, and can be applied to any expression dataset.
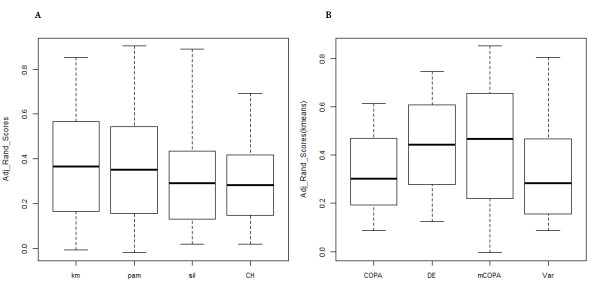
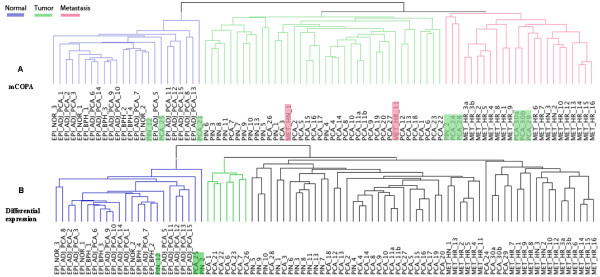
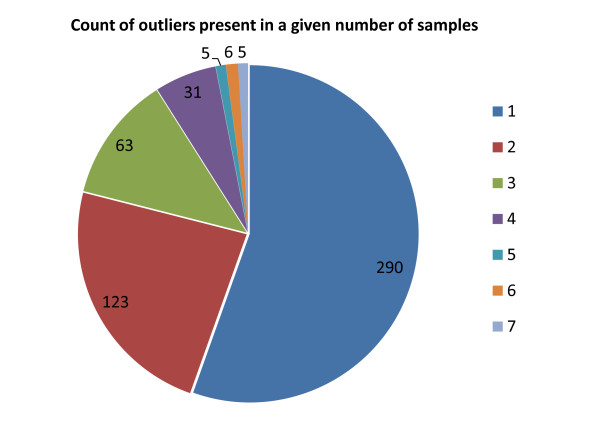
Results: We compare our method to other feature-selection approaches, and demonstrate that mCOPA frequently selects more-informative features than do differential expression or variance-based feature selection approaches, and is able to recover observed clinical subtypes more consistently. We demonstrate the application of mCOPA to prostate cancer expression data, and explore the use of outliers in clustering, pathway analysis, and the identification of tumour suppressors. We analyse the under-expressed outliers to identify known and novel prostate cancer tumour suppressor genes, validating these against data in Oncomine and the Cancer Gene Index. We also demonstrate how a combination of outlier analysis and pathway analysis can identify molecular mechanisms disrupted in individual tumours.
Conclusions: We demonstrate that mCOPA offers advantages, compared to differential expression or variance, in selecting outlier features, and that the features so selected are better able to assign samples to clinically annotated subtypes. Further, we show that the biology explored by outlier analysis differs from that uncovered in differential expression or variance analysis. mCOPA is an important new tool for the exploration of cancer datasets and the discovery of new cancer subtypes, and can be combined with pathway and functional analysis approaches to discover mechanisms underpinning heterogeneity in cancers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


