Andrew Stubbs, Elizabeth A McClellan, Sebastiaan Horsman, Saskia D Hiltemann, Ivo Palli, Stephan Nouwens, Anton Hj Koning, Frits Hoogland, Joke Reumers, Daphne Heijsman, Sigrid Swagemakers, Andreas Kremer, Jules Meijerink, Diether Lambrechts, Peter J van der Spek
{"title":"Huvariome:全基因组下一代等位基因频率测序的web服务器资源,以帮助病理候选基因选择。","authors":"Andrew Stubbs, Elizabeth A McClellan, Sebastiaan Horsman, Saskia D Hiltemann, Ivo Palli, Stephan Nouwens, Anton Hj Koning, Frits Hoogland, Joke Reumers, Daphne Heijsman, Sigrid Swagemakers, Andreas Kremer, Jules Meijerink, Diether Lambrechts, Peter J van der Spek","doi":"10.1186/2043-9113-2-19","DOIUrl":null,"url":null,"abstract":"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>Next generation sequencing provides clinical research scientists with direct read out of innumerable variants, including personal, pathological and common benign variants. The aim of resequencing studies is to determine the candidate pathogenic variants from individual genomes, or from family-based or tumor/normal genome comparisons. Whilst the use of appropriate controls within the experimental design will minimize the number of false positive variations selected, this number can be reduced further with the use of high quality whole genome reference data to minimize false positives variants prior to candidate gene selection. In addition the use of platform related sequencing error models can help in the recovery of ambiguous genotypes from lower coverage data.</p><p><strong>Description: </strong>We have developed a whole genome database of human genetic variations, Huvariome, determined by whole genome deep sequencing data with high coverage and low error rates. The database was designed to be sequencing technology independent but is currently populated with 165 individual whole genomes consisting of small pedigrees and matched tumor/normal samples sequenced with the Complete Genomics sequencing platform. Common variants have been determined for a Benelux population cohort and represented as genotypes alongside the results of two sets of control data (73 of the 165 genomes), Huvariome Core which comprises 31 healthy individuals from the Benelux region, and Diversity Panel consisting of 46 healthy individuals representing 10 different populations and 21 samples in three Pedigrees. Users can query the database by gene or position via a web interface and the results are displayed as the frequency of the variations as detected in the datasets. We demonstrate that Huvariome can provide accurate reference allele frequencies to disambiguate sequencing inconsistencies produced in resequencing experiments. Huvariome has been used to support the selection of candidate cardiomyopathy related genes which have a homozygous genotype in the reference cohorts. This database allows the users to see which selected variants are common variants (> 5% minor allele frequency) in the Huvariome core samples, thus aiding in the selection of potentially pathogenic variants by filtering out common variants that are not listed in one of the other public genomic variation databases. The no-call rate and the accuracy of allele calling in Huvariome provides the user with the possibility of identifying platform dependent errors associated with specific regions of the human genome.</p><p><strong>Conclusion: </strong>Huvariome is a simple to use resource for validation of resequencing results obtained by NGS experiments. The high sequence coverage and low error rates provide scientists with the ability to remove false positive results from pedigree studies. Results are returned via a web interface that displays location-based genetic variation frequency, impact on protein function, association with known genetic variations and a quality score of the variation base derived from Huvariome Core and the Diversity Panel data. These results may be used to identify and prioritize rare variants that, for example, might be disease relevant. In testing the accuracy of the Huvariome database, alleles of a selection of ambiguously called coding single nucleotide variants were successfully predicted in all cases. Data protection of individuals is ensured by restricted access to patient derived genomes from the host institution which is relevant for future molecular diagnostics.</p>","PeriodicalId":73663,"journal":{"name":"Journal of clinical bioinformatics","volume":"2 1","pages":"19"},"PeriodicalIF":0.0000,"publicationDate":"2012-11-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2043-9113-2-19","citationCount":"20","resultStr":"{\"title\":\"Huvariome: a web server resource of whole genome next-generation sequencing allelic frequencies to aid in pathological candidate gene selection.\",\"authors\":\"Andrew Stubbs, Elizabeth A McClellan, Sebastiaan Horsman, Saskia D Hiltemann, Ivo Palli, Stephan Nouwens, Anton Hj Koning, Frits Hoogland, Joke Reumers, Daphne Heijsman, Sigrid Swagemakers, Andreas Kremer, Jules Meijerink, Diether Lambrechts, Peter J van der Spek\",\"doi\":\"10.1186/2043-9113-2-19\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>Next generation sequencing provides clinical research scientists with direct read out of innumerable variants, including personal, pathological and common benign variants. The aim of resequencing studies is to determine the candidate pathogenic variants from individual genomes, or from family-based or tumor/normal genome comparisons. Whilst the use of appropriate controls within the experimental design will minimize the number of false positive variations selected, this number can be reduced further with the use of high quality whole genome reference data to minimize false positives variants prior to candidate gene selection. In addition the use of platform related sequencing error models can help in the recovery of ambiguous genotypes from lower coverage data.</p><p><strong>Description: </strong>We have developed a whole genome database of human genetic variations, Huvariome, determined by whole genome deep sequencing data with high coverage and low error rates. The database was designed to be sequencing technology independent but is currently populated with 165 individual whole genomes consisting of small pedigrees and matched tumor/normal samples sequenced with the Complete Genomics sequencing platform. Common variants have been determined for a Benelux population cohort and represented as genotypes alongside the results of two sets of control data (73 of the 165 genomes), Huvariome Core which comprises 31 healthy individuals from the Benelux region, and Diversity Panel consisting of 46 healthy individuals representing 10 different populations and 21 samples in three Pedigrees. Users can query the database by gene or position via a web interface and the results are displayed as the frequency of the variations as detected in the datasets. We demonstrate that Huvariome can provide accurate reference allele frequencies to disambiguate sequencing inconsistencies produced in resequencing experiments. Huvariome has been used to support the selection of candidate cardiomyopathy related genes which have a homozygous genotype in the reference cohorts. This database allows the users to see which selected variants are common variants (> 5% minor allele frequency) in the Huvariome core samples, thus aiding in the selection of potentially pathogenic variants by filtering out common variants that are not listed in one of the other public genomic variation databases. The no-call rate and the accuracy of allele calling in Huvariome provides the user with the possibility of identifying platform dependent errors associated with specific regions of the human genome.</p><p><strong>Conclusion: </strong>Huvariome is a simple to use resource for validation of resequencing results obtained by NGS experiments. The high sequence coverage and low error rates provide scientists with the ability to remove false positive results from pedigree studies. Results are returned via a web interface that displays location-based genetic variation frequency, impact on protein function, association with known genetic variations and a quality score of the variation base derived from Huvariome Core and the Diversity Panel data. These results may be used to identify and prioritize rare variants that, for example, might be disease relevant. In testing the accuracy of the Huvariome database, alleles of a selection of ambiguously called coding single nucleotide variants were successfully predicted in all cases. Data protection of individuals is ensured by restricted access to patient derived genomes from the host institution which is relevant for future molecular diagnostics.</p>\",\"PeriodicalId\":73663,\"journal\":{\"name\":\"Journal of clinical bioinformatics\",\"volume\":\"2 1\",\"pages\":\"19\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-11-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2043-9113-2-19\",\"citationCount\":\"20\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of clinical bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2043-9113-2-19\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2043-9113-2-19","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Huvariome: a web server resource of whole genome next-generation sequencing allelic frequencies to aid in pathological candidate gene selection.
Unlabelled:
Background: Next generation sequencing provides clinical research scientists with direct read out of innumerable variants, including personal, pathological and common benign variants. The aim of resequencing studies is to determine the candidate pathogenic variants from individual genomes, or from family-based or tumor/normal genome comparisons. Whilst the use of appropriate controls within the experimental design will minimize the number of false positive variations selected, this number can be reduced further with the use of high quality whole genome reference data to minimize false positives variants prior to candidate gene selection. In addition the use of platform related sequencing error models can help in the recovery of ambiguous genotypes from lower coverage data.
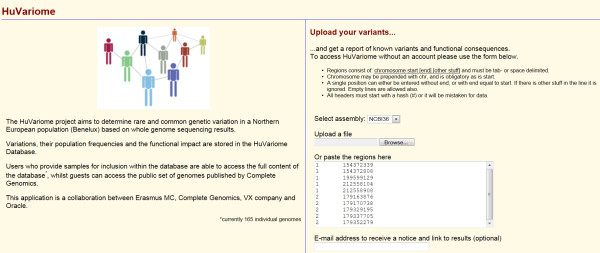
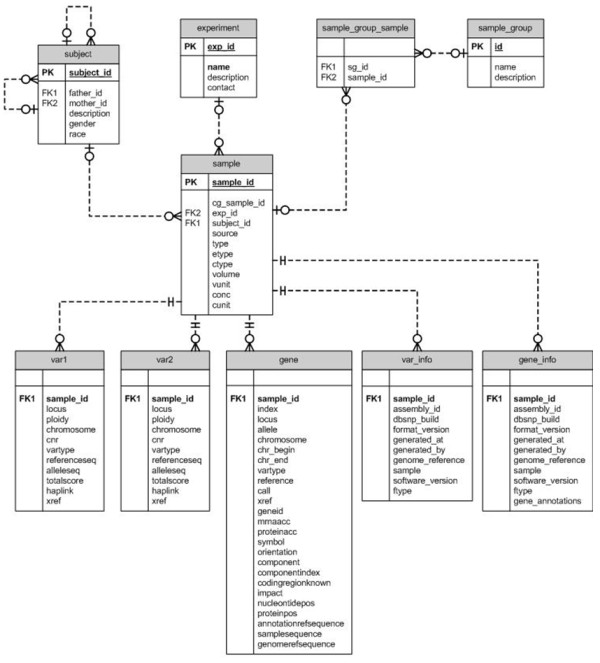
Description: We have developed a whole genome database of human genetic variations, Huvariome, determined by whole genome deep sequencing data with high coverage and low error rates. The database was designed to be sequencing technology independent but is currently populated with 165 individual whole genomes consisting of small pedigrees and matched tumor/normal samples sequenced with the Complete Genomics sequencing platform. Common variants have been determined for a Benelux population cohort and represented as genotypes alongside the results of two sets of control data (73 of the 165 genomes), Huvariome Core which comprises 31 healthy individuals from the Benelux region, and Diversity Panel consisting of 46 healthy individuals representing 10 different populations and 21 samples in three Pedigrees. Users can query the database by gene or position via a web interface and the results are displayed as the frequency of the variations as detected in the datasets. We demonstrate that Huvariome can provide accurate reference allele frequencies to disambiguate sequencing inconsistencies produced in resequencing experiments. Huvariome has been used to support the selection of candidate cardiomyopathy related genes which have a homozygous genotype in the reference cohorts. This database allows the users to see which selected variants are common variants (> 5% minor allele frequency) in the Huvariome core samples, thus aiding in the selection of potentially pathogenic variants by filtering out common variants that are not listed in one of the other public genomic variation databases. The no-call rate and the accuracy of allele calling in Huvariome provides the user with the possibility of identifying platform dependent errors associated with specific regions of the human genome.
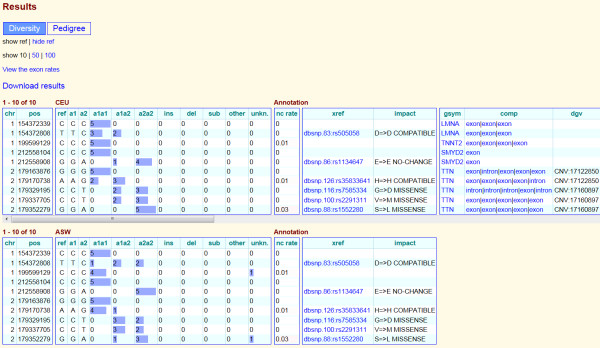
Conclusion: Huvariome is a simple to use resource for validation of resequencing results obtained by NGS experiments. The high sequence coverage and low error rates provide scientists with the ability to remove false positive results from pedigree studies. Results are returned via a web interface that displays location-based genetic variation frequency, impact on protein function, association with known genetic variations and a quality score of the variation base derived from Huvariome Core and the Diversity Panel data. These results may be used to identify and prioritize rare variants that, for example, might be disease relevant. In testing the accuracy of the Huvariome database, alleles of a selection of ambiguously called coding single nucleotide variants were successfully predicted in all cases. Data protection of individuals is ensured by restricted access to patient derived genomes from the host institution which is relevant for future molecular diagnostics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


