Stéphane Deschamps, Kishore Nannapaneni, Yun Zhang, Kevin Hayes
{"title":"用illumina基因组分析仪对玉米配对端简化表示文库的局部片段进行测序。","authors":"Stéphane Deschamps, Kishore Nannapaneni, Yun Zhang, Kevin Hayes","doi":"10.1155/2012/360598","DOIUrl":null,"url":null,"abstract":"<p><p>The use of next-generation DNA sequencing technologies has greatly facilitated reference-guided variant detection in complex plant genomes. However, complications may arise when regions adjacent to a read of interest are used for marker assay development, or when reference sequences are incomplete, as short reads alone may not be long enough to ascertain their uniqueness. Here, the possibility of generating longer sequences in discrete regions of the large and complex genome of maize is demonstrated, using a modified version of a paired-end RAD library construction strategy. Reads are generated from DNA fragments first digested with a methylation-sensitive restriction endonuclease, sheared, enriched with biotin and a selective PCR amplification step, and then sequenced at both ends. Sequences are locally assembled into contigs by subgrouping pairs based on the identity of the read anchored by the restriction site. This strategy applied to two maize inbred lines (B14 and B73) generated 183,609 and 129,018 contigs, respectively, out of which at least 76% were >200 bps in length. A subset of putative single nucleotide polymorphisms from contigs aligning to the B73 reference genome with at least one mismatch was resequenced, and 90% of those in B14 were confirmed, indicating that this method is a potent approach for variant detection and marker development in species with complex genomes or lacking extensive reference sequences.</p>","PeriodicalId":73471,"journal":{"name":"International journal of plant genomics","volume":"2012 ","pages":"360598"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2012/360598","citationCount":"6","resultStr":"{\"title\":\"Local assemblies of paired-end reduced representation libraries sequenced with the illumina genome analyzer in maize.\",\"authors\":\"Stéphane Deschamps, Kishore Nannapaneni, Yun Zhang, Kevin Hayes\",\"doi\":\"10.1155/2012/360598\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The use of next-generation DNA sequencing technologies has greatly facilitated reference-guided variant detection in complex plant genomes. However, complications may arise when regions adjacent to a read of interest are used for marker assay development, or when reference sequences are incomplete, as short reads alone may not be long enough to ascertain their uniqueness. Here, the possibility of generating longer sequences in discrete regions of the large and complex genome of maize is demonstrated, using a modified version of a paired-end RAD library construction strategy. Reads are generated from DNA fragments first digested with a methylation-sensitive restriction endonuclease, sheared, enriched with biotin and a selective PCR amplification step, and then sequenced at both ends. Sequences are locally assembled into contigs by subgrouping pairs based on the identity of the read anchored by the restriction site. This strategy applied to two maize inbred lines (B14 and B73) generated 183,609 and 129,018 contigs, respectively, out of which at least 76% were >200 bps in length. A subset of putative single nucleotide polymorphisms from contigs aligning to the B73 reference genome with at least one mismatch was resequenced, and 90% of those in B14 were confirmed, indicating that this method is a potent approach for variant detection and marker development in species with complex genomes or lacking extensive reference sequences.</p>\",\"PeriodicalId\":73471,\"journal\":{\"name\":\"International journal of plant genomics\",\"volume\":\"2012 \",\"pages\":\"360598\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2012/360598\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International journal of plant genomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2012/360598\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2012/10/9 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International journal of plant genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2012/360598","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2012/10/9 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Local assemblies of paired-end reduced representation libraries sequenced with the illumina genome analyzer in maize.
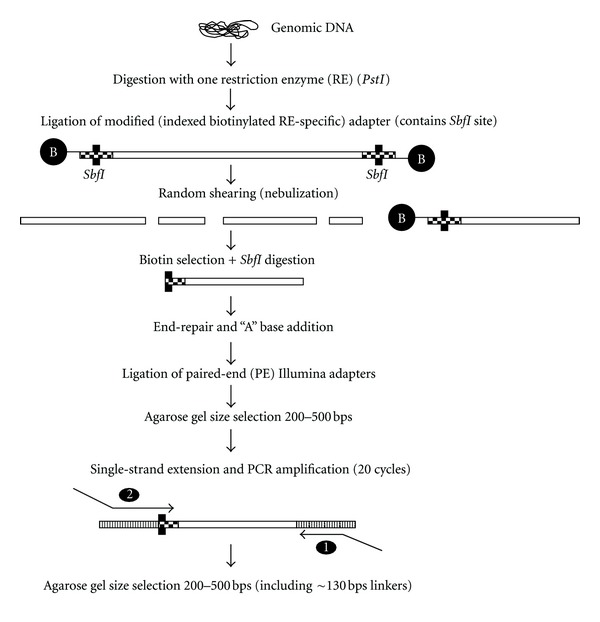
The use of next-generation DNA sequencing technologies has greatly facilitated reference-guided variant detection in complex plant genomes. However, complications may arise when regions adjacent to a read of interest are used for marker assay development, or when reference sequences are incomplete, as short reads alone may not be long enough to ascertain their uniqueness. Here, the possibility of generating longer sequences in discrete regions of the large and complex genome of maize is demonstrated, using a modified version of a paired-end RAD library construction strategy. Reads are generated from DNA fragments first digested with a methylation-sensitive restriction endonuclease, sheared, enriched with biotin and a selective PCR amplification step, and then sequenced at both ends. Sequences are locally assembled into contigs by subgrouping pairs based on the identity of the read anchored by the restriction site. This strategy applied to two maize inbred lines (B14 and B73) generated 183,609 and 129,018 contigs, respectively, out of which at least 76% were >200 bps in length. A subset of putative single nucleotide polymorphisms from contigs aligning to the B73 reference genome with at least one mismatch was resequenced, and 90% of those in B14 were confirmed, indicating that this method is a potent approach for variant detection and marker development in species with complex genomes or lacking extensive reference sequences.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


