Alexey B Mantsyzov, Guillaume Bouvier, Nathalie Evrard-Todeschi, Gildas Bertho
{"title":"基于接触的配体聚类方法在虚拟筛选中鉴定活性化合物。","authors":"Alexey B Mantsyzov, Guillaume Bouvier, Nathalie Evrard-Todeschi, Gildas Bertho","doi":"10.2147/AABC.S30881","DOIUrl":null,"url":null,"abstract":"<p><p>Evaluation of docking results is one of the most important problems for virtual screening and in silico drug design. Modern approaches for the identification of active compounds in a large data set of docked molecules use energy scoring functions. One of the general and most significant limitations of these methods relates to inaccurate binding energy estimation, which results in false scoring of docked compounds. Automatic analysis of poses using self-organizing maps (AuPosSOM) represents an alternative approach for the evaluation of docking results based on the clustering of compounds by the similarity of their contacts with the receptor. A scoring function was developed for the identification of the active compounds in the AuPosSOM clustered dataset. In addition, the AuPosSOM efficiency for the clustering of compounds and the identification of key contacts considered as important for its activity, were also improved. Benchmark tests for several targets revealed that together with the developed scoring function, AuPosSOM represents a good alternative to the energy-based scoring functions for the evaluation of docking results.</p>","PeriodicalId":53584,"journal":{"name":"Advances and Applications in Bioinformatics and Chemistry","volume":"5 ","pages":"61-79"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/AABC.S30881","citationCount":"26","resultStr":"{\"title\":\"Contact-based ligand-clustering approach for the identification of active compounds in virtual screening.\",\"authors\":\"Alexey B Mantsyzov, Guillaume Bouvier, Nathalie Evrard-Todeschi, Gildas Bertho\",\"doi\":\"10.2147/AABC.S30881\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Evaluation of docking results is one of the most important problems for virtual screening and in silico drug design. Modern approaches for the identification of active compounds in a large data set of docked molecules use energy scoring functions. One of the general and most significant limitations of these methods relates to inaccurate binding energy estimation, which results in false scoring of docked compounds. Automatic analysis of poses using self-organizing maps (AuPosSOM) represents an alternative approach for the evaluation of docking results based on the clustering of compounds by the similarity of their contacts with the receptor. A scoring function was developed for the identification of the active compounds in the AuPosSOM clustered dataset. In addition, the AuPosSOM efficiency for the clustering of compounds and the identification of key contacts considered as important for its activity, were also improved. Benchmark tests for several targets revealed that together with the developed scoring function, AuPosSOM represents a good alternative to the energy-based scoring functions for the evaluation of docking results.</p>\",\"PeriodicalId\":53584,\"journal\":{\"name\":\"Advances and Applications in Bioinformatics and Chemistry\",\"volume\":\"5 \",\"pages\":\"61-79\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.2147/AABC.S30881\",\"citationCount\":\"26\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Advances and Applications in Bioinformatics and Chemistry\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2147/AABC.S30881\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2012/9/6 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances and Applications in Bioinformatics and Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/AABC.S30881","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2012/9/6 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Contact-based ligand-clustering approach for the identification of active compounds in virtual screening.
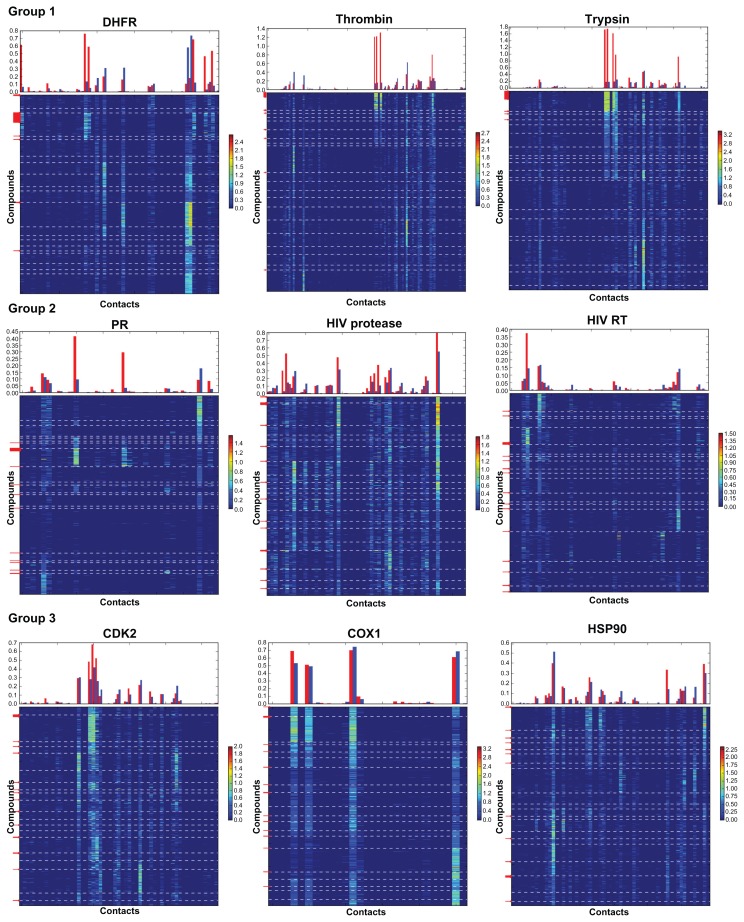
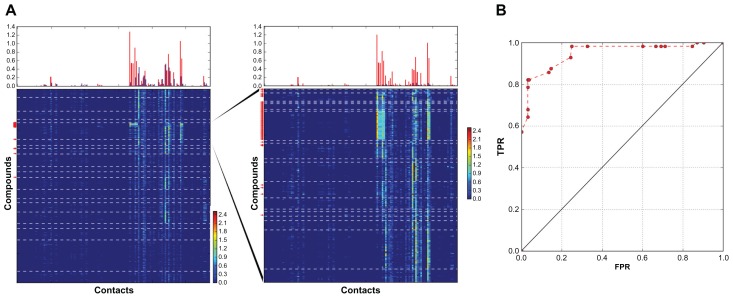
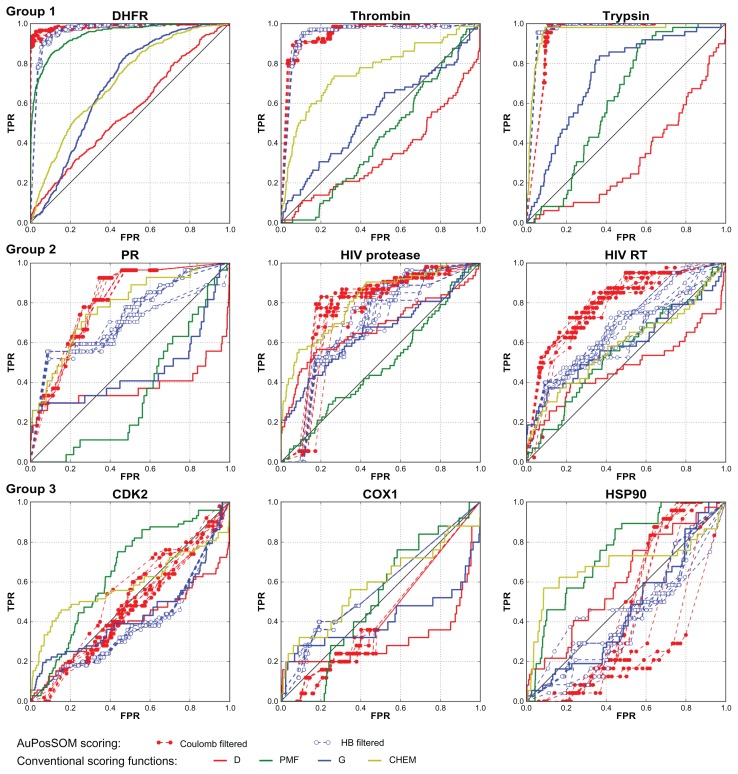
Evaluation of docking results is one of the most important problems for virtual screening and in silico drug design. Modern approaches for the identification of active compounds in a large data set of docked molecules use energy scoring functions. One of the general and most significant limitations of these methods relates to inaccurate binding energy estimation, which results in false scoring of docked compounds. Automatic analysis of poses using self-organizing maps (AuPosSOM) represents an alternative approach for the evaluation of docking results based on the clustering of compounds by the similarity of their contacts with the receptor. A scoring function was developed for the identification of the active compounds in the AuPosSOM clustered dataset. In addition, the AuPosSOM efficiency for the clustering of compounds and the identification of key contacts considered as important for its activity, were also improved. Benchmark tests for several targets revealed that together with the developed scoring function, AuPosSOM represents a good alternative to the energy-based scoring functions for the evaluation of docking results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


