{"title":"21个分枝杆菌基因组(结核和非结核菌株)的计算基因组学-蛋白质组学和系统发育分析。","authors":"Fathiah Zakham, Othmane Aouane, David Ussery, Abdelaziz Benjouad, Moulay Mustapha Ennaji","doi":"10.1186/2042-5783-2-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>The genus Mycobacterium comprises different species, among them the most contagious and infectious bacteria. The members of the complex Mycobacterium tuberculosis are the most virulent microorganisms that have killed human and other mammals since millennia. Additionally, with the many different mycobacterial sequences available, there is a crucial need for the visualization and the simplification of their data. In this present study, we aim to highlight a comparative genome, proteome and phylogeny analysis between twenty-one mycobacterial (Tuberculosis and non tuberculosis) strains using a set of computational and bioinformatics tools (Pan and Core genome plotting, BLAST matrix and phylogeny analysis).</p><p><strong>Results: </strong>Considerably the result of pan and core genome Plotting demonstrated that less than 1250 Mycobacterium gene families are conserved across all species, and a total set of about 20,000 gene families within the Mycobacterium pan-genome of twenty one mycobacterial genomes.Viewing the BLAST matrix a high similarity was found among the species of the complex Mycobacterium tuberculosis and less conservation is found with other slow growing pathogenic mycobacteria.Phylogeny analysis based on both protein conservation, as well as rRNA clearly resolve known relationships between slow growing mycobacteria.</p><p><strong>Conclusion: </strong>Mycobacteria include important pathogenic species for human and animals and the Mycobacterium tuberculosis complex is the most cause of death of the humankind. The comparative genome analysis could provide a new insight for better controlling and preventing these diseases.</p>","PeriodicalId":18538,"journal":{"name":"Microbial Informatics and Experimentation","volume":"2 1","pages":"7"},"PeriodicalIF":0.0000,"publicationDate":"2012-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2042-5783-2-7","citationCount":"28","resultStr":"{\"title\":\"Computational genomics-proteomics and Phylogeny analysis of twenty one mycobacterial genomes (Tuberculosis & non Tuberculosis strains).\",\"authors\":\"Fathiah Zakham, Othmane Aouane, David Ussery, Abdelaziz Benjouad, Moulay Mustapha Ennaji\",\"doi\":\"10.1186/2042-5783-2-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>The genus Mycobacterium comprises different species, among them the most contagious and infectious bacteria. The members of the complex Mycobacterium tuberculosis are the most virulent microorganisms that have killed human and other mammals since millennia. Additionally, with the many different mycobacterial sequences available, there is a crucial need for the visualization and the simplification of their data. In this present study, we aim to highlight a comparative genome, proteome and phylogeny analysis between twenty-one mycobacterial (Tuberculosis and non tuberculosis) strains using a set of computational and bioinformatics tools (Pan and Core genome plotting, BLAST matrix and phylogeny analysis).</p><p><strong>Results: </strong>Considerably the result of pan and core genome Plotting demonstrated that less than 1250 Mycobacterium gene families are conserved across all species, and a total set of about 20,000 gene families within the Mycobacterium pan-genome of twenty one mycobacterial genomes.Viewing the BLAST matrix a high similarity was found among the species of the complex Mycobacterium tuberculosis and less conservation is found with other slow growing pathogenic mycobacteria.Phylogeny analysis based on both protein conservation, as well as rRNA clearly resolve known relationships between slow growing mycobacteria.</p><p><strong>Conclusion: </strong>Mycobacteria include important pathogenic species for human and animals and the Mycobacterium tuberculosis complex is the most cause of death of the humankind. The comparative genome analysis could provide a new insight for better controlling and preventing these diseases.</p>\",\"PeriodicalId\":18538,\"journal\":{\"name\":\"Microbial Informatics and Experimentation\",\"volume\":\"2 1\",\"pages\":\"7\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-08-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2042-5783-2-7\",\"citationCount\":\"28\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microbial Informatics and Experimentation\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2042-5783-2-7\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Informatics and Experimentation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2042-5783-2-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Computational genomics-proteomics and Phylogeny analysis of twenty one mycobacterial genomes (Tuberculosis & non Tuberculosis strains).
Unlabelled:
Background: The genus Mycobacterium comprises different species, among them the most contagious and infectious bacteria. The members of the complex Mycobacterium tuberculosis are the most virulent microorganisms that have killed human and other mammals since millennia. Additionally, with the many different mycobacterial sequences available, there is a crucial need for the visualization and the simplification of their data. In this present study, we aim to highlight a comparative genome, proteome and phylogeny analysis between twenty-one mycobacterial (Tuberculosis and non tuberculosis) strains using a set of computational and bioinformatics tools (Pan and Core genome plotting, BLAST matrix and phylogeny analysis).
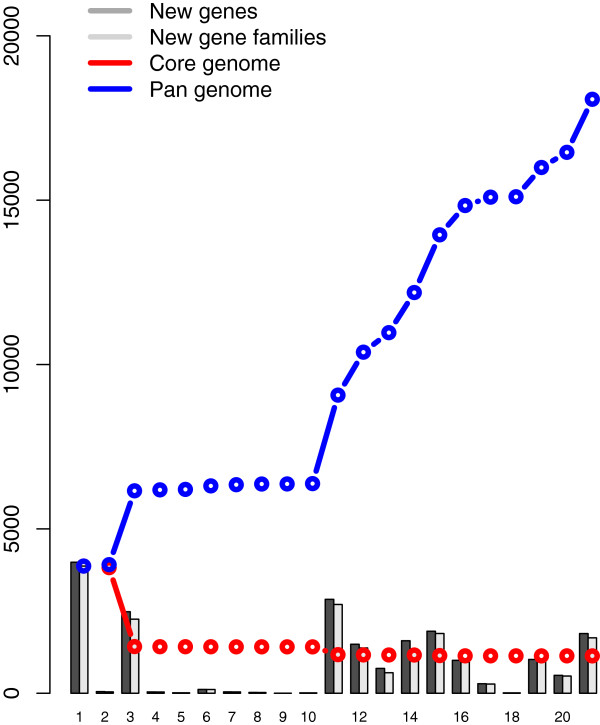
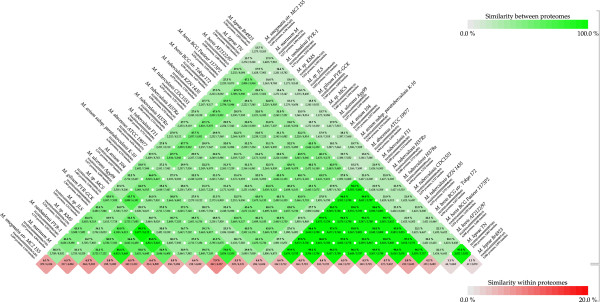
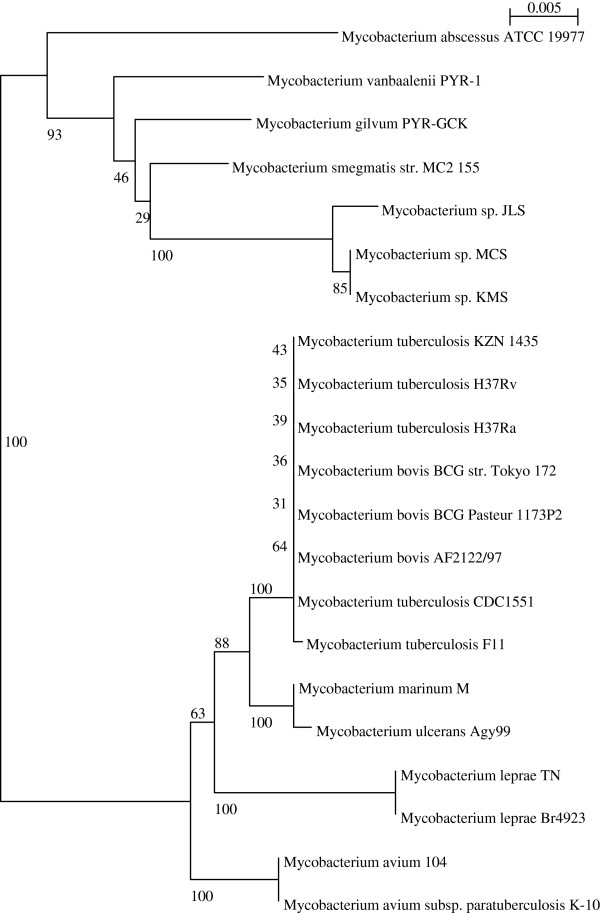
Results: Considerably the result of pan and core genome Plotting demonstrated that less than 1250 Mycobacterium gene families are conserved across all species, and a total set of about 20,000 gene families within the Mycobacterium pan-genome of twenty one mycobacterial genomes.Viewing the BLAST matrix a high similarity was found among the species of the complex Mycobacterium tuberculosis and less conservation is found with other slow growing pathogenic mycobacteria.Phylogeny analysis based on both protein conservation, as well as rRNA clearly resolve known relationships between slow growing mycobacteria.
Conclusion: Mycobacteria include important pathogenic species for human and animals and the Mycobacterium tuberculosis complex is the most cause of death of the humankind. The comparative genome analysis could provide a new insight for better controlling and preventing these diseases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


