Jan Hasenauer, Julian Heinrich, Malgorzata Doszczak, Peter Scheurich, Daniel Weiskopf, Frank Allgöwer
{"title":"异质细胞群模型的可视化分析方法。","authors":"Jan Hasenauer, Julian Heinrich, Malgorzata Doszczak, Peter Scheurich, Daniel Weiskopf, Frank Allgöwer","doi":"10.1186/1687-4153-2012-4","DOIUrl":null,"url":null,"abstract":"<p><p> In recent years, cell population models have become increasingly common. In contrast to classic single cell models, population models allow for the study of cell-to-cell variability, a crucial phenomenon in most populations of primary cells, cancer cells, and stem cells. Unfortunately, tools for in-depth analysis of population models are still missing. This problem originates from the complexity of population models. Particularly important are methods to determine the source of heterogeneity (e.g., genetics or epigenetic differences) and to select potential (bio-)markers. We propose an analysis based on visual analytics to tackle this problem. Our approach combines parallel-coordinates plots, used for a visual assessment of the high-dimensional dependencies, and nonlinear support vector machines, for the quantification of effects. The method can be employed to study qualitative and quantitative differences among cells. To illustrate the different components, we perform a case study using the proapoptotic signal transduction pathway involved in cellular apoptosis.</p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2012 1","pages":"4"},"PeriodicalIF":0.0000,"publicationDate":"2012-05-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1687-4153-2012-4","citationCount":"12","resultStr":"{\"title\":\"A visual analytics approach for models of heterogeneous cell populations.\",\"authors\":\"Jan Hasenauer, Julian Heinrich, Malgorzata Doszczak, Peter Scheurich, Daniel Weiskopf, Frank Allgöwer\",\"doi\":\"10.1186/1687-4153-2012-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p> In recent years, cell population models have become increasingly common. In contrast to classic single cell models, population models allow for the study of cell-to-cell variability, a crucial phenomenon in most populations of primary cells, cancer cells, and stem cells. Unfortunately, tools for in-depth analysis of population models are still missing. This problem originates from the complexity of population models. Particularly important are methods to determine the source of heterogeneity (e.g., genetics or epigenetic differences) and to select potential (bio-)markers. We propose an analysis based on visual analytics to tackle this problem. Our approach combines parallel-coordinates plots, used for a visual assessment of the high-dimensional dependencies, and nonlinear support vector machines, for the quantification of effects. The method can be employed to study qualitative and quantitative differences among cells. To illustrate the different components, we perform a case study using the proapoptotic signal transduction pathway involved in cellular apoptosis.</p>\",\"PeriodicalId\":72957,\"journal\":{\"name\":\"EURASIP journal on bioinformatics & systems biology\",\"volume\":\"2012 1\",\"pages\":\"4\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-05-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1687-4153-2012-4\",\"citationCount\":\"12\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EURASIP journal on bioinformatics & systems biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1687-4153-2012-4\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1687-4153-2012-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
A visual analytics approach for models of heterogeneous cell populations.
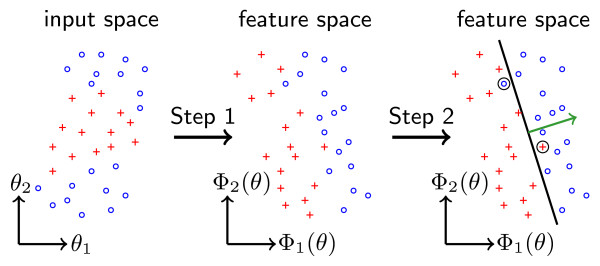
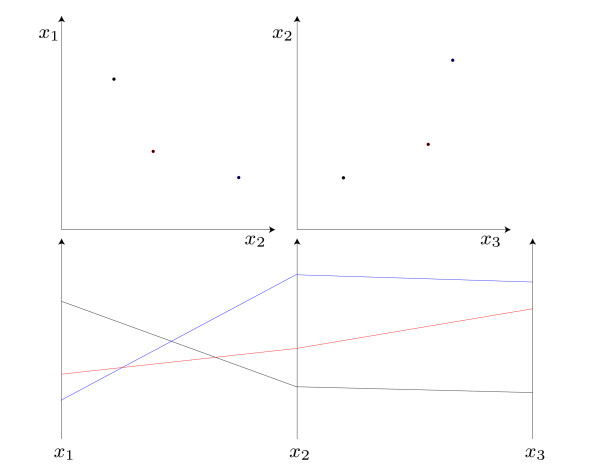
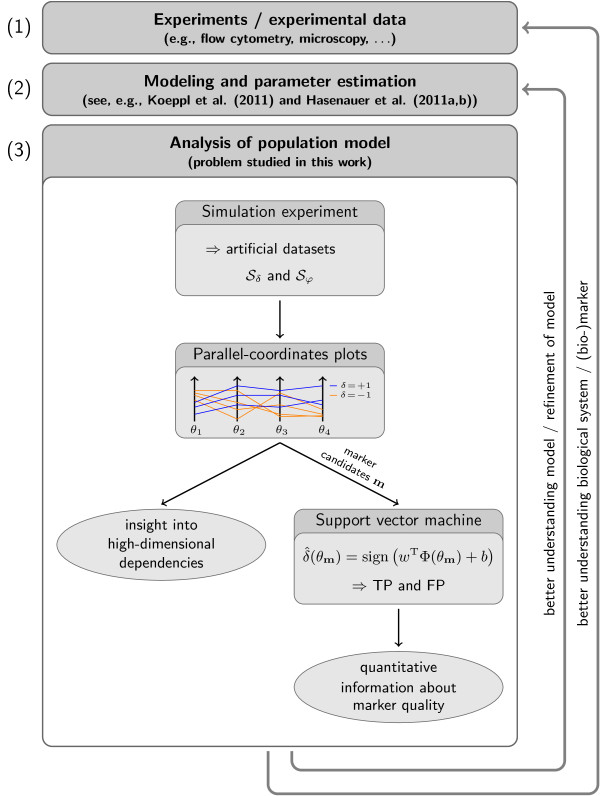
In recent years, cell population models have become increasingly common. In contrast to classic single cell models, population models allow for the study of cell-to-cell variability, a crucial phenomenon in most populations of primary cells, cancer cells, and stem cells. Unfortunately, tools for in-depth analysis of population models are still missing. This problem originates from the complexity of population models. Particularly important are methods to determine the source of heterogeneity (e.g., genetics or epigenetic differences) and to select potential (bio-)markers. We propose an analysis based on visual analytics to tackle this problem. Our approach combines parallel-coordinates plots, used for a visual assessment of the high-dimensional dependencies, and nonlinear support vector machines, for the quantification of effects. The method can be employed to study qualitative and quantitative differences among cells. To illustrate the different components, we perform a case study using the proapoptotic signal transduction pathway involved in cellular apoptosis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


