Manal Helal, Fanrong Kong, Sharon Ca Chen, Fei Zhou, Dominic E Dwyer, John Potter, Vitali Sintchenko
{"title":"线性归一化哈希函数用于基因序列聚类和从多个序列比对中识别参考序列。","authors":"Manal Helal, Fanrong Kong, Sharon Ca Chen, Fei Zhou, Dominic E Dwyer, John Potter, Vitali Sintchenko","doi":"10.1186/2042-5783-2-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Comparative genomics has put additional demands on the assessment of similarity between sequences and their clustering as means for classification. However, defining the optimal number of clusters, cluster density and boundaries for sets of potentially related sequences of genes with variable degrees of polymorphism remains a significant challenge. The aim of this study was to develop a method that would identify the cluster centroids and the optimal number of clusters for a given sensitivity level and could work equally well for the different sequence datasets.</p><p><strong>Results: </strong>A novel method that combines the linear mapping hash function and multiple sequence alignment (MSA) was developed. This method takes advantage of the already sorted by similarity sequences from the MSA output, and identifies the optimal number of clusters, clusters cut-offs, and clusters centroids that can represent reference gene vouchers for the different species. The linear mapping hash function can map an already ordered by similarity distance matrix to indices to reveal gaps in the values around which the optimal cut-offs of the different clusters can be identified. The method was evaluated using sets of closely related (16S rRNA gene sequences of Nocardia species) and highly variable (VP1 genomic region of Enterovirus 71) sequences and outperformed existing unsupervised machine learning clustering methods and dimensionality reduction methods. This method does not require prior knowledge of the number of clusters or the distance between clusters, handles clusters of different sizes and shapes, and scales linearly with the dataset.</p><p><strong>Conclusions: </strong>The combination of MSA with the linear mapping hash function is a computationally efficient way of gene sequence clustering and can be a valuable tool for the assessment of similarity, clustering of different microbial genomes, identifying reference sequences, and for the study of evolution of bacteria and viruses.</p>","PeriodicalId":18538,"journal":{"name":"Microbial Informatics and Experimentation","volume":"2 1","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2042-5783-2-2","citationCount":"2","resultStr":"{\"title\":\"Linear normalised hash function for clustering gene sequences and identifying reference sequences from multiple sequence alignments.\",\"authors\":\"Manal Helal, Fanrong Kong, Sharon Ca Chen, Fei Zhou, Dominic E Dwyer, John Potter, Vitali Sintchenko\",\"doi\":\"10.1186/2042-5783-2-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Comparative genomics has put additional demands on the assessment of similarity between sequences and their clustering as means for classification. However, defining the optimal number of clusters, cluster density and boundaries for sets of potentially related sequences of genes with variable degrees of polymorphism remains a significant challenge. The aim of this study was to develop a method that would identify the cluster centroids and the optimal number of clusters for a given sensitivity level and could work equally well for the different sequence datasets.</p><p><strong>Results: </strong>A novel method that combines the linear mapping hash function and multiple sequence alignment (MSA) was developed. This method takes advantage of the already sorted by similarity sequences from the MSA output, and identifies the optimal number of clusters, clusters cut-offs, and clusters centroids that can represent reference gene vouchers for the different species. The linear mapping hash function can map an already ordered by similarity distance matrix to indices to reveal gaps in the values around which the optimal cut-offs of the different clusters can be identified. The method was evaluated using sets of closely related (16S rRNA gene sequences of Nocardia species) and highly variable (VP1 genomic region of Enterovirus 71) sequences and outperformed existing unsupervised machine learning clustering methods and dimensionality reduction methods. This method does not require prior knowledge of the number of clusters or the distance between clusters, handles clusters of different sizes and shapes, and scales linearly with the dataset.</p><p><strong>Conclusions: </strong>The combination of MSA with the linear mapping hash function is a computationally efficient way of gene sequence clustering and can be a valuable tool for the assessment of similarity, clustering of different microbial genomes, identifying reference sequences, and for the study of evolution of bacteria and viruses.</p>\",\"PeriodicalId\":18538,\"journal\":{\"name\":\"Microbial Informatics and Experimentation\",\"volume\":\"2 1\",\"pages\":\"2\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-01-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2042-5783-2-2\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microbial Informatics and Experimentation\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2042-5783-2-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Informatics and Experimentation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2042-5783-2-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Linear normalised hash function for clustering gene sequences and identifying reference sequences from multiple sequence alignments.
Background: Comparative genomics has put additional demands on the assessment of similarity between sequences and their clustering as means for classification. However, defining the optimal number of clusters, cluster density and boundaries for sets of potentially related sequences of genes with variable degrees of polymorphism remains a significant challenge. The aim of this study was to develop a method that would identify the cluster centroids and the optimal number of clusters for a given sensitivity level and could work equally well for the different sequence datasets.
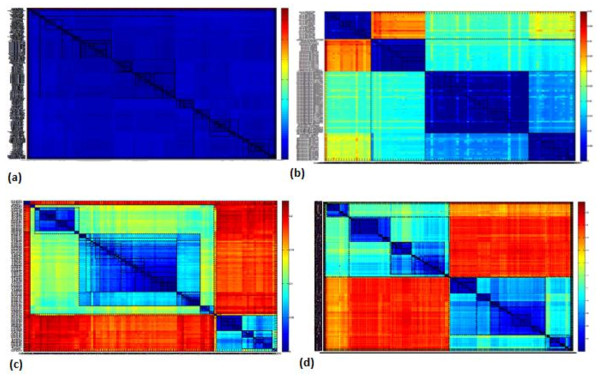
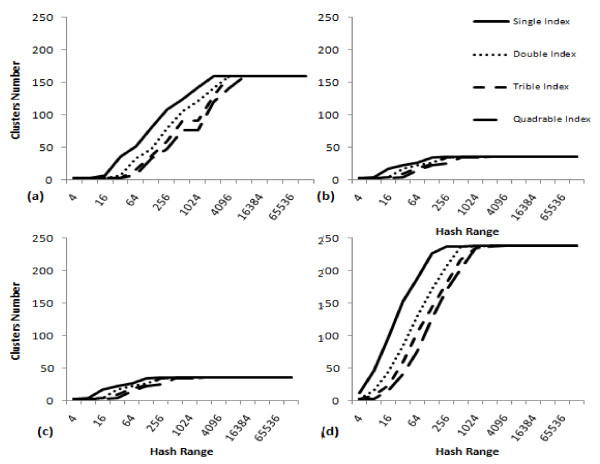
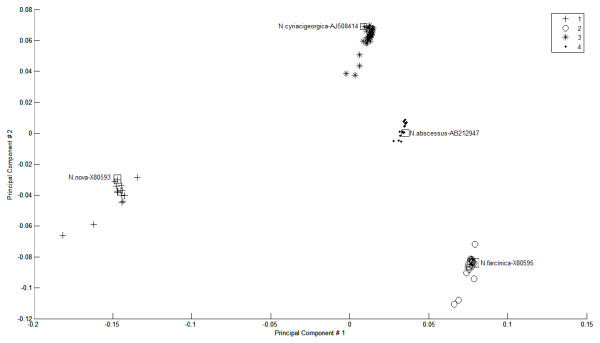
Results: A novel method that combines the linear mapping hash function and multiple sequence alignment (MSA) was developed. This method takes advantage of the already sorted by similarity sequences from the MSA output, and identifies the optimal number of clusters, clusters cut-offs, and clusters centroids that can represent reference gene vouchers for the different species. The linear mapping hash function can map an already ordered by similarity distance matrix to indices to reveal gaps in the values around which the optimal cut-offs of the different clusters can be identified. The method was evaluated using sets of closely related (16S rRNA gene sequences of Nocardia species) and highly variable (VP1 genomic region of Enterovirus 71) sequences and outperformed existing unsupervised machine learning clustering methods and dimensionality reduction methods. This method does not require prior knowledge of the number of clusters or the distance between clusters, handles clusters of different sizes and shapes, and scales linearly with the dataset.
Conclusions: The combination of MSA with the linear mapping hash function is a computationally efficient way of gene sequence clustering and can be a valuable tool for the assessment of similarity, clustering of different microbial genomes, identifying reference sequences, and for the study of evolution of bacteria and viruses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


