{"title":"基于对接模型的异源二聚体界面分类及复杂结构预测的评分函数构建。","authors":"Yuko Tsuchiya, Eiji Kanamori, Haruki Nakamura, Kengo Kinoshita","doi":"10.2147/aabc.s6347","DOIUrl":null,"url":null,"abstract":"<p><p>Protein-protein docking simulations can provide the predicted complex structural models. In a docking simulation, several putative structural models are selected by scoring functions from an ensemble of many complex models. Scoring functions based on statistical analyses of heterodimers are usually designed to select the complex model with the most abundant interaction mode found among the known complexes, as the correct model. However, because the formation schemes of heterodimers are extremely diverse, a single scoring function does not seem to be sufficient to describe the fitness of the predicted models other than the most abundant interaction mode. Thus, it is necessary to classify the heterodimers in terms of their individual interaction modes, and then to construct multiple scoring functions for each heterodimer type. In this study, we constructed the classification method of heterodimers based on the discriminative characters between near-native and decoy models, which were found in the comparison of the interfaces in terms of the complementarities for the hydrophobicity, the electrostatic potential and the shape. Consequently, we found four heterodimer clusters, and then constructed the multiple scoring functions, each of which was optimized for each cluster. Our multiple scoring functions were applied to the predictions in the unbound docking.</p>","PeriodicalId":53584,"journal":{"name":"Advances and Applications in Bioinformatics and Chemistry","volume":"2 ","pages":"79-100"},"PeriodicalIF":0.0000,"publicationDate":"2009-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/aabc.s6347","citationCount":"2","resultStr":"{\"title\":\"Classification of heterodimer interfaces using docking models and construction of scoring functions for the complex structure prediction.\",\"authors\":\"Yuko Tsuchiya, Eiji Kanamori, Haruki Nakamura, Kengo Kinoshita\",\"doi\":\"10.2147/aabc.s6347\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Protein-protein docking simulations can provide the predicted complex structural models. In a docking simulation, several putative structural models are selected by scoring functions from an ensemble of many complex models. Scoring functions based on statistical analyses of heterodimers are usually designed to select the complex model with the most abundant interaction mode found among the known complexes, as the correct model. However, because the formation schemes of heterodimers are extremely diverse, a single scoring function does not seem to be sufficient to describe the fitness of the predicted models other than the most abundant interaction mode. Thus, it is necessary to classify the heterodimers in terms of their individual interaction modes, and then to construct multiple scoring functions for each heterodimer type. In this study, we constructed the classification method of heterodimers based on the discriminative characters between near-native and decoy models, which were found in the comparison of the interfaces in terms of the complementarities for the hydrophobicity, the electrostatic potential and the shape. Consequently, we found four heterodimer clusters, and then constructed the multiple scoring functions, each of which was optimized for each cluster. Our multiple scoring functions were applied to the predictions in the unbound docking.</p>\",\"PeriodicalId\":53584,\"journal\":{\"name\":\"Advances and Applications in Bioinformatics and Chemistry\",\"volume\":\"2 \",\"pages\":\"79-100\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2009-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.2147/aabc.s6347\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Advances and Applications in Bioinformatics and Chemistry\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2147/aabc.s6347\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2009/9/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances and Applications in Bioinformatics and Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/aabc.s6347","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2009/9/22 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Classification of heterodimer interfaces using docking models and construction of scoring functions for the complex structure prediction.
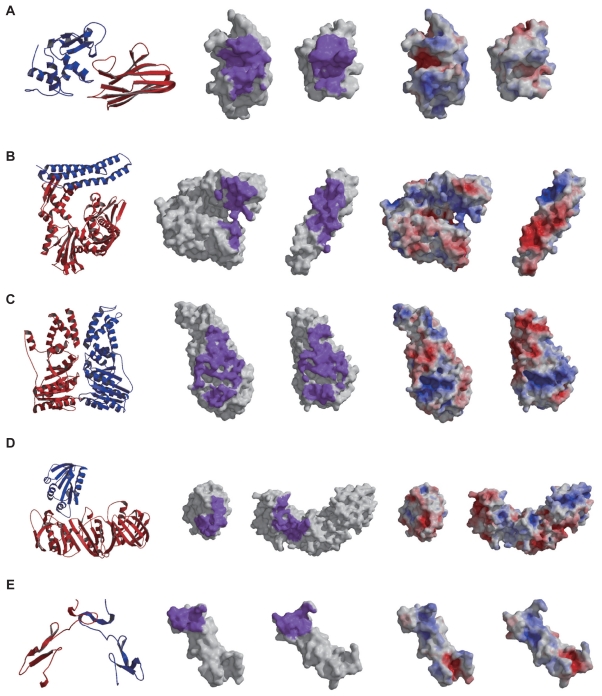
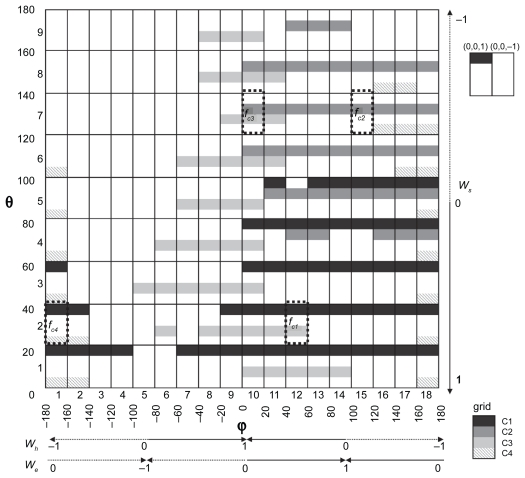
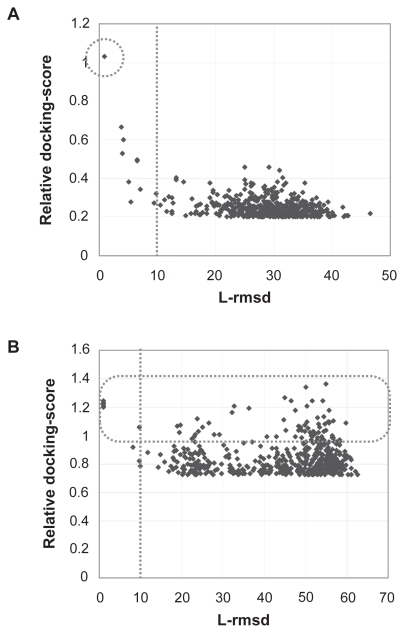
Protein-protein docking simulations can provide the predicted complex structural models. In a docking simulation, several putative structural models are selected by scoring functions from an ensemble of many complex models. Scoring functions based on statistical analyses of heterodimers are usually designed to select the complex model with the most abundant interaction mode found among the known complexes, as the correct model. However, because the formation schemes of heterodimers are extremely diverse, a single scoring function does not seem to be sufficient to describe the fitness of the predicted models other than the most abundant interaction mode. Thus, it is necessary to classify the heterodimers in terms of their individual interaction modes, and then to construct multiple scoring functions for each heterodimer type. In this study, we constructed the classification method of heterodimers based on the discriminative characters between near-native and decoy models, which were found in the comparison of the interfaces in terms of the complementarities for the hydrophobicity, the electrostatic potential and the shape. Consequently, we found four heterodimer clusters, and then constructed the multiple scoring functions, each of which was optimized for each cluster. Our multiple scoring functions were applied to the predictions in the unbound docking.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


