Rui-Ru Ji, Karl-Heinz Ott, Roumyana Yordanova, Robert E Bruccoleri
{"title":"FDR-FET:一种优化基因集富集分析方法。","authors":"Rui-Ru Ji, Karl-Heinz Ott, Roumyana Yordanova, Robert E Bruccoleri","doi":"10.2147/AABC.S15840","DOIUrl":null,"url":null,"abstract":"<p><p>Gene set enrichment analysis for analyzing large profiling and screening experiments can reveal unifying biological schemes based on previously accumulated knowledge represented as \"gene sets\". Most of the existing implementations use a fixed fold-change or P value cutoff to generate regulated gene lists. However, the threshold selection in most cases is arbitrary, and has a significant effect on the test outcome and interpretation of the experiment. We developed a new gene set enrichment analysis method, ie, FDR-FET, which dynamically optimizes the threshold choice and improves the sensitivity and selectivity of gene set enrichment analysis. The procedure translates experimental results into a series of regulated gene lists at multiple false discovery rate (FDR) cutoffs, and computes the P value of the overrepresentation of a gene set using a Fisher's exact test (FET) in each of these gene lists. The lowest P value is retained to represent the significance of the gene set. We also implemented improved methods to define a more relevant global reference set for the FET. We demonstrate the validity of the method using a published microarray study of three protease inhibitors of the human immunodeficiency virus and compare the results with those from other popular gene set enrichment analysis algorithms. Our results show that combining FDR with multiple cutoffs allows us to control the error while retaining genes that increase information content. We conclude that FDR-FET can selectively identify significant affected biological processes. Our method can be used for any user-generated gene list in the area of transcriptome, proteome, and other biological and scientific applications.</p>","PeriodicalId":53584,"journal":{"name":"Advances and Applications in Bioinformatics and Chemistry","volume":"4 ","pages":"37-42"},"PeriodicalIF":0.0000,"publicationDate":"2011-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/AABC.S15840","citationCount":"3","resultStr":"{\"title\":\"FDR-FET: an optimizing gene set enrichment analysis method.\",\"authors\":\"Rui-Ru Ji, Karl-Heinz Ott, Roumyana Yordanova, Robert E Bruccoleri\",\"doi\":\"10.2147/AABC.S15840\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Gene set enrichment analysis for analyzing large profiling and screening experiments can reveal unifying biological schemes based on previously accumulated knowledge represented as \\\"gene sets\\\". Most of the existing implementations use a fixed fold-change or P value cutoff to generate regulated gene lists. However, the threshold selection in most cases is arbitrary, and has a significant effect on the test outcome and interpretation of the experiment. We developed a new gene set enrichment analysis method, ie, FDR-FET, which dynamically optimizes the threshold choice and improves the sensitivity and selectivity of gene set enrichment analysis. The procedure translates experimental results into a series of regulated gene lists at multiple false discovery rate (FDR) cutoffs, and computes the P value of the overrepresentation of a gene set using a Fisher's exact test (FET) in each of these gene lists. The lowest P value is retained to represent the significance of the gene set. We also implemented improved methods to define a more relevant global reference set for the FET. We demonstrate the validity of the method using a published microarray study of three protease inhibitors of the human immunodeficiency virus and compare the results with those from other popular gene set enrichment analysis algorithms. Our results show that combining FDR with multiple cutoffs allows us to control the error while retaining genes that increase information content. We conclude that FDR-FET can selectively identify significant affected biological processes. Our method can be used for any user-generated gene list in the area of transcriptome, proteome, and other biological and scientific applications.</p>\",\"PeriodicalId\":53584,\"journal\":{\"name\":\"Advances and Applications in Bioinformatics and Chemistry\",\"volume\":\"4 \",\"pages\":\"37-42\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2011-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.2147/AABC.S15840\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Advances and Applications in Bioinformatics and Chemistry\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2147/AABC.S15840\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2011/3/15 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances and Applications in Bioinformatics and Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/AABC.S15840","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2011/3/15 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
FDR-FET: an optimizing gene set enrichment analysis method.
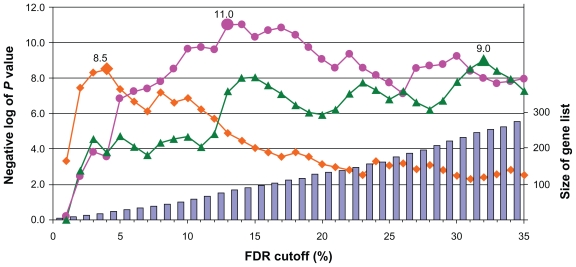
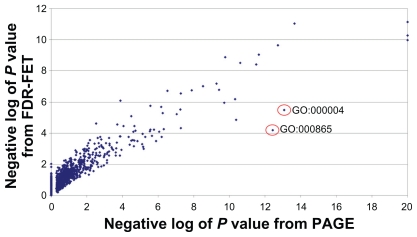
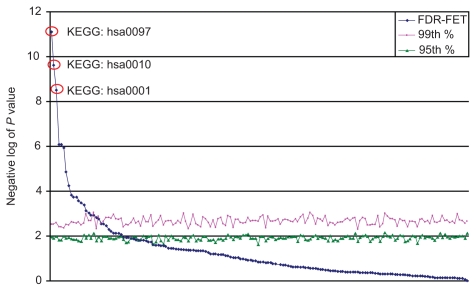
Gene set enrichment analysis for analyzing large profiling and screening experiments can reveal unifying biological schemes based on previously accumulated knowledge represented as "gene sets". Most of the existing implementations use a fixed fold-change or P value cutoff to generate regulated gene lists. However, the threshold selection in most cases is arbitrary, and has a significant effect on the test outcome and interpretation of the experiment. We developed a new gene set enrichment analysis method, ie, FDR-FET, which dynamically optimizes the threshold choice and improves the sensitivity and selectivity of gene set enrichment analysis. The procedure translates experimental results into a series of regulated gene lists at multiple false discovery rate (FDR) cutoffs, and computes the P value of the overrepresentation of a gene set using a Fisher's exact test (FET) in each of these gene lists. The lowest P value is retained to represent the significance of the gene set. We also implemented improved methods to define a more relevant global reference set for the FET. We demonstrate the validity of the method using a published microarray study of three protease inhibitors of the human immunodeficiency virus and compare the results with those from other popular gene set enrichment analysis algorithms. Our results show that combining FDR with multiple cutoffs allows us to control the error while retaining genes that increase information content. We conclude that FDR-FET can selectively identify significant affected biological processes. Our method can be used for any user-generated gene list in the area of transcriptome, proteome, and other biological and scientific applications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


