{"title":"利用序列比对约束对带有假结的rna进行有效比对。","authors":"Byung-Jun Yoon","doi":"10.1155/2009/491074","DOIUrl":null,"url":null,"abstract":"<p><p>When aligning RNAs, it is important to consider both the secondary structure similarity and primary sequence similarity to find an accurate alignment. However, algorithms that can handle RNA secondary structures typically have high computational complexity that limits their utility. For this reason, there have been a number of attempts to find useful alignment constraints that can reduce the computations without sacrificing the alignment accuracy. In this paper, we propose a new method for finding effective alignment constraints for fast and accurate structural alignment of RNAs, including pseudoknots. In the proposed method, we use a profile-HMM to identify the \"seed\" regions that can be aligned with high confidence. We also estimate the position range of the aligned bases that are located outside the seed regions. The location of the seed regions and the estimated range of the alignment positions are then used to establish the sequence alignment constraints. We incorporated the proposed constraints into the profile context-sensitive HMM (profile-csHMM) based RNA structural alignment algorithm. Experiments indicate that the proposed method can make the alignment speed up to 11 times faster without degrading the accuracy of the RNA alignment.</p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":" ","pages":"491074"},"PeriodicalIF":0.0000,"publicationDate":"2009-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2009/491074","citationCount":"5","resultStr":"{\"title\":\"Efficient alignment of RNAs with pseudoknots using sequence alignment constraints.\",\"authors\":\"Byung-Jun Yoon\",\"doi\":\"10.1155/2009/491074\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>When aligning RNAs, it is important to consider both the secondary structure similarity and primary sequence similarity to find an accurate alignment. However, algorithms that can handle RNA secondary structures typically have high computational complexity that limits their utility. For this reason, there have been a number of attempts to find useful alignment constraints that can reduce the computations without sacrificing the alignment accuracy. In this paper, we propose a new method for finding effective alignment constraints for fast and accurate structural alignment of RNAs, including pseudoknots. In the proposed method, we use a profile-HMM to identify the \\\"seed\\\" regions that can be aligned with high confidence. We also estimate the position range of the aligned bases that are located outside the seed regions. The location of the seed regions and the estimated range of the alignment positions are then used to establish the sequence alignment constraints. We incorporated the proposed constraints into the profile context-sensitive HMM (profile-csHMM) based RNA structural alignment algorithm. Experiments indicate that the proposed method can make the alignment speed up to 11 times faster without degrading the accuracy of the RNA alignment.</p>\",\"PeriodicalId\":72957,\"journal\":{\"name\":\"EURASIP journal on bioinformatics & systems biology\",\"volume\":\" \",\"pages\":\"491074\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2009-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2009/491074\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EURASIP journal on bioinformatics & systems biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2009/491074\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2009/4/14 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2009/491074","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2009/4/14 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Efficient alignment of RNAs with pseudoknots using sequence alignment constraints.
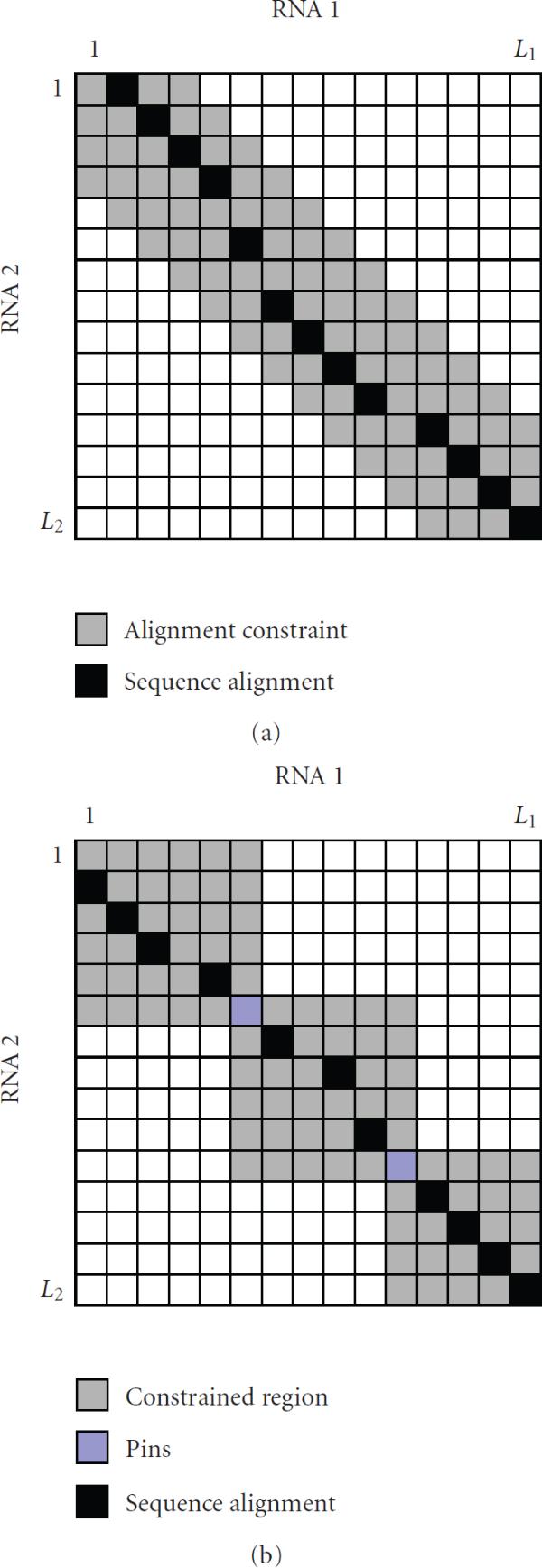
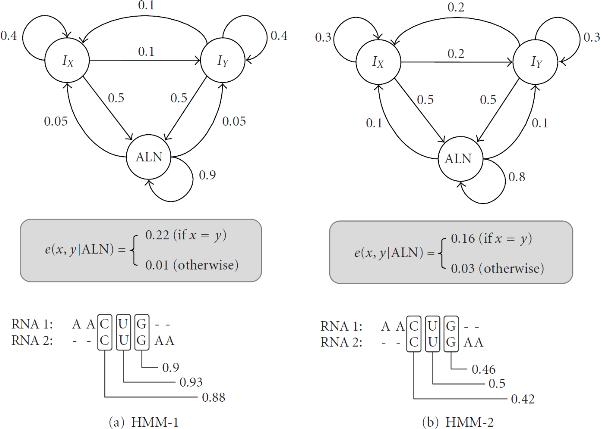
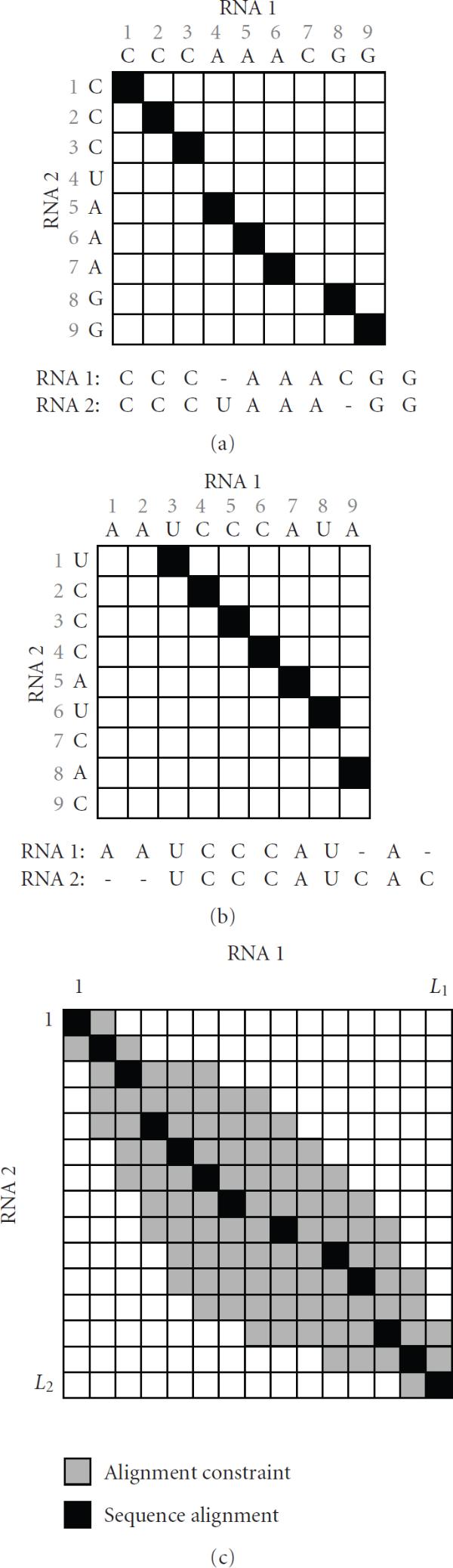
When aligning RNAs, it is important to consider both the secondary structure similarity and primary sequence similarity to find an accurate alignment. However, algorithms that can handle RNA secondary structures typically have high computational complexity that limits their utility. For this reason, there have been a number of attempts to find useful alignment constraints that can reduce the computations without sacrificing the alignment accuracy. In this paper, we propose a new method for finding effective alignment constraints for fast and accurate structural alignment of RNAs, including pseudoknots. In the proposed method, we use a profile-HMM to identify the "seed" regions that can be aligned with high confidence. We also estimate the position range of the aligned bases that are located outside the seed regions. The location of the seed regions and the estimated range of the alignment positions are then used to establish the sequence alignment constraints. We incorporated the proposed constraints into the profile context-sensitive HMM (profile-csHMM) based RNA structural alignment algorithm. Experiments indicate that the proposed method can make the alignment speed up to 11 times faster without degrading the accuracy of the RNA alignment.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


