{"title":"С6H6和1,3,5- ad3c6h3与CBr3反应的DFT机理研究。氢化物从芳香族CH键转移到亲电试剂的第一个例子","authors":"Yurii A. Borisov, Irena S. Akhrem","doi":"10.1016/j.molcata.2016.10.027","DOIUrl":null,"url":null,"abstract":"<div><p>The DFT B3LYP/6-31G* calculations were carried out for the reactions of C<sub>6</sub>H<sub>6</sub> and Ad<sub>3</sub>C<sub>6</sub>H<sub>3</sub> (Ad<!--> <!-->=<!--> <!-->1,3,5-adamantyl) with superelectrophile CBr<sub>3</sub><sup>+</sup> as a model of superelectrophilic catalyst CBr<sub>3</sub><sup>+</sup> Al<sub>2</sub>Br<sub>7</sub><sup>−</sup>. The reaction of C<sub>6</sub>H<sub>6</sub> with CBr<sub>3</sub><sup>+</sup> proceeds <em>via</em> the classical scheme of electrophilic reactions of aromatic C<!--> <!-->−<!--> <!-->H bond to form initially the barrier-free σ-complex C<sub>6</sub>H<sub>6</sub>CBr<sub>3</sub><sup>+</sup>. This mechanism was confirmed by the aug-cc-pVDZ basis set calculations. The reaction of Ad<sub>3</sub>C<sub>6</sub>H<sub>3</sub> with CBr<sub>3</sub><sup>+</sup> occurs <em>via</em> a quite novel mechanism involving aryl cation formation followed by hydride abstraction of the Ar<sup>+</sup> from the 2-Ad group and the rearrangement of the 2-Ad<sup>+</sup> cation into the 4-phenyl-4-protoadamantyl cation. The hydride transfer from both arenes was shown to be more favorable than H radical transfer by more than 40 and 55<!--> <!-->kcal<!--> <!-->mol<sup>−1</sup> in the case of C<sub>6</sub>H<sub>6</sub> and Ad<sub>3</sub>C<sub>6</sub>H<sub>3</sub>, respectively.</p></div>","PeriodicalId":370,"journal":{"name":"Journal of Molecular Catalysis A: Chemical","volume":"426 ","pages":"Pages 610-617"},"PeriodicalIF":5.0620,"publicationDate":"2017-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.molcata.2016.10.027","citationCount":"5","resultStr":"{\"title\":\"DFT mechanistic study of reactions of С6H6 and 1,3,5-Ad3C6H3 with CBr3. The first example of hydride transfer from aromatic CH bond to electrophile\",\"authors\":\"Yurii A. Borisov, Irena S. Akhrem\",\"doi\":\"10.1016/j.molcata.2016.10.027\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The DFT B3LYP/6-31G* calculations were carried out for the reactions of C<sub>6</sub>H<sub>6</sub> and Ad<sub>3</sub>C<sub>6</sub>H<sub>3</sub> (Ad<!--> <!-->=<!--> <!-->1,3,5-adamantyl) with superelectrophile CBr<sub>3</sub><sup>+</sup> as a model of superelectrophilic catalyst CBr<sub>3</sub><sup>+</sup> Al<sub>2</sub>Br<sub>7</sub><sup>−</sup>. The reaction of C<sub>6</sub>H<sub>6</sub> with CBr<sub>3</sub><sup>+</sup> proceeds <em>via</em> the classical scheme of electrophilic reactions of aromatic C<!--> <!-->−<!--> <!-->H bond to form initially the barrier-free σ-complex C<sub>6</sub>H<sub>6</sub>CBr<sub>3</sub><sup>+</sup>. This mechanism was confirmed by the aug-cc-pVDZ basis set calculations. The reaction of Ad<sub>3</sub>C<sub>6</sub>H<sub>3</sub> with CBr<sub>3</sub><sup>+</sup> occurs <em>via</em> a quite novel mechanism involving aryl cation formation followed by hydride abstraction of the Ar<sup>+</sup> from the 2-Ad group and the rearrangement of the 2-Ad<sup>+</sup> cation into the 4-phenyl-4-protoadamantyl cation. The hydride transfer from both arenes was shown to be more favorable than H radical transfer by more than 40 and 55<!--> <!-->kcal<!--> <!-->mol<sup>−1</sup> in the case of C<sub>6</sub>H<sub>6</sub> and Ad<sub>3</sub>C<sub>6</sub>H<sub>3</sub>, respectively.</p></div>\",\"PeriodicalId\":370,\"journal\":{\"name\":\"Journal of Molecular Catalysis A: Chemical\",\"volume\":\"426 \",\"pages\":\"Pages 610-617\"},\"PeriodicalIF\":5.0620,\"publicationDate\":\"2017-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1016/j.molcata.2016.10.027\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Catalysis A: Chemical\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1381116916304496\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Catalysis A: Chemical","FirstCategoryId":"1","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1381116916304496","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 5
摘要
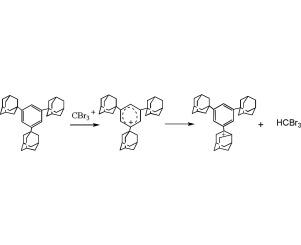
以超亲电催化剂CBr3+ Al2Br7−为模型,对C6H6和Ad3C6H3 (Ad = 1,3,5-adamantyl)与超亲电试剂CBr3+的反应进行了DFT B3LYP/6-31G*计算。C6H6与CBr3+的反应遵循芳香族C−H键亲电反应的经典模式,初步形成无障碍的σ-配合物C6H6CBr3+。该机理得到了aug-cc-pVDZ基集计算的证实。Ad3C6H3与CBr3+的反应发生在芳基阳离子形成后,2-Ad基上的Ar+被氢化物萃取,2-Ad+阳离子重排为4-苯基-4-原金刚烷基阳离子。结果表明,C6H6和Ad3C6H3的氢化物转移比H自由基转移更有利,转移量分别大于40和55 kcal mol−1。
DFT mechanistic study of reactions of С6H6 and 1,3,5-Ad3C6H3 with CBr3. The first example of hydride transfer from aromatic CH bond to electrophile
The DFT B3LYP/6-31G* calculations were carried out for the reactions of C6H6 and Ad3C6H3 (Ad = 1,3,5-adamantyl) with superelectrophile CBr3+ as a model of superelectrophilic catalyst CBr3+ Al2Br7−. The reaction of C6H6 with CBr3+ proceeds via the classical scheme of electrophilic reactions of aromatic C − H bond to form initially the barrier-free σ-complex C6H6CBr3+. This mechanism was confirmed by the aug-cc-pVDZ basis set calculations. The reaction of Ad3C6H3 with CBr3+ occurs via a quite novel mechanism involving aryl cation formation followed by hydride abstraction of the Ar+ from the 2-Ad group and the rearrangement of the 2-Ad+ cation into the 4-phenyl-4-protoadamantyl cation. The hydride transfer from both arenes was shown to be more favorable than H radical transfer by more than 40 and 55 kcal mol−1 in the case of C6H6 and Ad3C6H3, respectively.
期刊介绍:
The Journal of Molecular Catalysis A: Chemical publishes original, rigorous, and scholarly full papers that examine the molecular and atomic aspects of catalytic activation and reaction mechanisms in homogeneous catalysis, heterogeneous catalysis (including supported organometallic catalysis), and computational catalysis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


