Laura Uelze, Josephine Grützke, Maria Borowiak, Jens Andre Hammerl, Katharina Juraschek, Carlus Deneke, Simon H Tausch, Burkhard Malorny
{"title":"基于全基因组测序数据的分型方法。","authors":"Laura Uelze, Josephine Grützke, Maria Borowiak, Jens Andre Hammerl, Katharina Juraschek, Carlus Deneke, Simon H Tausch, Burkhard Malorny","doi":"10.1186/s42522-020-0010-1","DOIUrl":null,"url":null,"abstract":"<p><p>Whole genome sequencing (WGS) of foodborne pathogens has become an effective method for investigating the information contained in the genome sequence of bacterial pathogens. In addition, its highly discriminative power enables the comparison of genetic relatedness between bacteria even on a sub-species level. For this reason, WGS is being implemented worldwide and across sectors (human, veterinary, food, and environment) for the investigation of disease outbreaks, source attribution, and improved risk characterization models. In order to extract relevant information from the large quantity and complex data produced by WGS, a host of bioinformatics tools has been developed, allowing users to analyze and interpret sequencing data, starting from simple gene-searches to complex phylogenetic studies. Depending on the research question, the complexity of the dataset and their bioinformatics skill set, users can choose between a great variety of tools for the analysis of WGS data. In this review, we describe the relevant approaches for phylogenomic studies for outbreak studies and give an overview of selected tools for the characterization of foodborne pathogens based on WGS data. Despite the efforts of the last years, harmonization and standardization of typing tools are still urgently needed to allow for an easy comparison of data between laboratories, moving towards a one health worldwide surveillance system for foodborne pathogens.</p>","PeriodicalId":19490,"journal":{"name":"One Health Outlook","volume":"2 ","pages":"3"},"PeriodicalIF":0.0000,"publicationDate":"2020-02-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7993478/pdf/","citationCount":"0","resultStr":"{\"title\":\"Typing methods based on whole genome sequencing data.\",\"authors\":\"Laura Uelze, Josephine Grützke, Maria Borowiak, Jens Andre Hammerl, Katharina Juraschek, Carlus Deneke, Simon H Tausch, Burkhard Malorny\",\"doi\":\"10.1186/s42522-020-0010-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Whole genome sequencing (WGS) of foodborne pathogens has become an effective method for investigating the information contained in the genome sequence of bacterial pathogens. In addition, its highly discriminative power enables the comparison of genetic relatedness between bacteria even on a sub-species level. For this reason, WGS is being implemented worldwide and across sectors (human, veterinary, food, and environment) for the investigation of disease outbreaks, source attribution, and improved risk characterization models. In order to extract relevant information from the large quantity and complex data produced by WGS, a host of bioinformatics tools has been developed, allowing users to analyze and interpret sequencing data, starting from simple gene-searches to complex phylogenetic studies. Depending on the research question, the complexity of the dataset and their bioinformatics skill set, users can choose between a great variety of tools for the analysis of WGS data. In this review, we describe the relevant approaches for phylogenomic studies for outbreak studies and give an overview of selected tools for the characterization of foodborne pathogens based on WGS data. Despite the efforts of the last years, harmonization and standardization of typing tools are still urgently needed to allow for an easy comparison of data between laboratories, moving towards a one health worldwide surveillance system for foodborne pathogens.</p>\",\"PeriodicalId\":19490,\"journal\":{\"name\":\"One Health Outlook\",\"volume\":\"2 \",\"pages\":\"3\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-02-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7993478/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"One Health Outlook\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s42522-020-0010-1\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"One Health Outlook","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42522-020-0010-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Typing methods based on whole genome sequencing data.
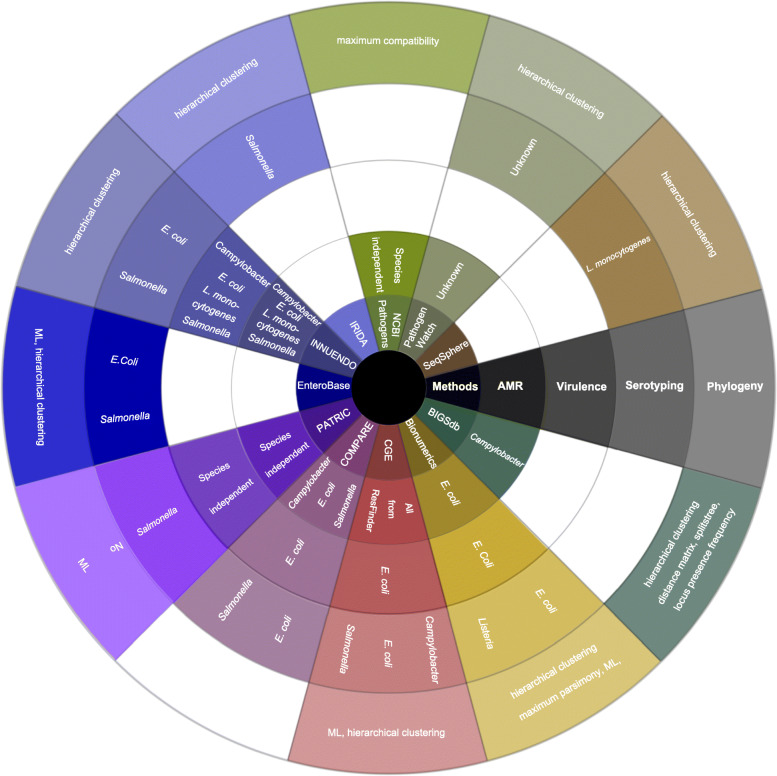
Whole genome sequencing (WGS) of foodborne pathogens has become an effective method for investigating the information contained in the genome sequence of bacterial pathogens. In addition, its highly discriminative power enables the comparison of genetic relatedness between bacteria even on a sub-species level. For this reason, WGS is being implemented worldwide and across sectors (human, veterinary, food, and environment) for the investigation of disease outbreaks, source attribution, and improved risk characterization models. In order to extract relevant information from the large quantity and complex data produced by WGS, a host of bioinformatics tools has been developed, allowing users to analyze and interpret sequencing data, starting from simple gene-searches to complex phylogenetic studies. Depending on the research question, the complexity of the dataset and their bioinformatics skill set, users can choose between a great variety of tools for the analysis of WGS data. In this review, we describe the relevant approaches for phylogenomic studies for outbreak studies and give an overview of selected tools for the characterization of foodborne pathogens based on WGS data. Despite the efforts of the last years, harmonization and standardization of typing tools are still urgently needed to allow for an easy comparison of data between laboratories, moving towards a one health worldwide surveillance system for foodborne pathogens.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


