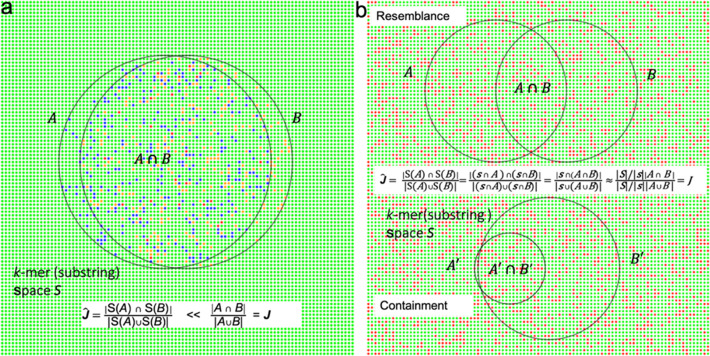
Kssd:通过 k-mer 子串空间采样降低序列维度,实现实时大规模数据集分析。
IF 12.3
1区 生物学
Q1 Agricultural and Biological Sciences
引用次数: 0
摘要
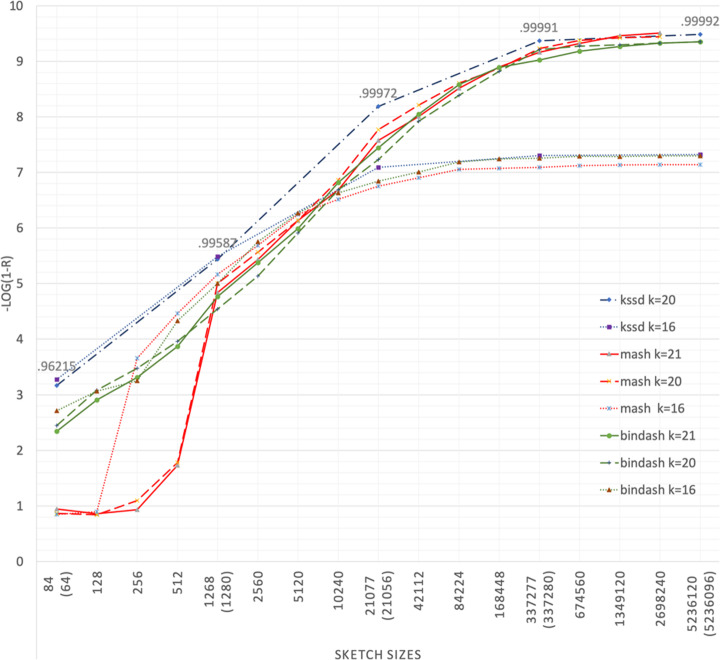
在这里,我们开发了 k -mer 子串空间分解(Kssd),这是一种草图绘制技术,与当前的草图绘制方法相比,速度更快,精度更高。我们的研究表明,这是唯一一种可以在模拟数据和真实数据的群体分辨率下进行大规模数据集比较的方法。利用 Kssd,我们对 NCBI Sequence Read Archive 中所有 1,019,179 个细菌全基因组测序(WGS)运行的参考文献进行了优先排序,发现其中 6164 个存在识别错误或污染。此外,我们还分析了来自 1000 基因组计划的 WGS 和外显子组运行样本。本文章由计算机程序翻译,如有差异,请以英文原文为准。

Kssd: sequence dimensionality reduction by k-mer substring space sampling enables real-time large-scale datasets analysis.
Here, we develop k -mer substring space decomposition (Kssd), a sketching technique which is significantly faster and more accurate than current sketching methods. We show that it is the only method that can be used for large-scale dataset comparisons at population resolution on simulated and real data. Using Kssd, we prioritize references for all 1,019,179 bacteria whole genome sequencing (WGS) runs from NCBI Sequence Read Archive and find misidentification or contamination in 6164 of these. Additionally, we analyze WGS and exome runs of samples from the 1000 Genomes Project.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Genome Biology
BIOTECHNOLOGY & APPLIED MICROBIOLOGY-GENETICS & HEREDITY
CiteScore
25.50
自引率
3.30%
发文量
0
审稿时长
14 weeks
期刊介绍:
Genome Biology is a leading research journal that focuses on the study of biology and biomedicine from a genomic and post-genomic standpoint. The journal consistently publishes outstanding research across various areas within these fields.
With an impressive impact factor of 12.3 (2022), Genome Biology has earned its place as the 3rd highest-ranked research journal in the Genetics and Heredity category, according to Thomson Reuters. Additionally, it is ranked 2nd among research journals in the Biotechnology and Applied Microbiology category. It is important to note that Genome Biology is the top-ranking open access journal in this category.
In summary, Genome Biology sets a high standard for scientific publications in the field, showcasing cutting-edge research and earning recognition among its peers.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


