Matthew T Parker, Katarzyna Knop, Geoffrey J Barton, Gordon G Simpson
{"title":"2passstools:使用机器学习过滤剪接连接的两遍比对提高了长读RNA测序中内含子检测的准确性。","authors":"Matthew T Parker, Katarzyna Knop, Geoffrey J Barton, Gordon G Simpson","doi":"10.1186/s13059-021-02296-0","DOIUrl":null,"url":null,"abstract":"<p><p>Transcription of eukaryotic genomes involves complex alternative processing of RNAs. Sequencing of full-length RNAs using long reads reveals the true complexity of processing. However, the relatively high error rates of long-read sequencing technologies can reduce the accuracy of intron identification. Here we apply alignment metrics and machine-learning-derived sequence information to filter spurious splice junctions from long-read alignments and use the remaining junctions to guide realignment in a two-pass approach. This method, available in the software package 2passtools ( https://github.com/bartongroup/2passtools ), improves the accuracy of spliced alignment and transcriptome assembly for species both with and without existing high-quality annotations.</p>","PeriodicalId":48922,"journal":{"name":"Genome Biology","volume":"22 1","pages":"72"},"PeriodicalIF":12.3000,"publicationDate":"2021-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13059-021-02296-0","citationCount":"13","resultStr":"{\"title\":\"2passtools: two-pass alignment using machine-learning-filtered splice junctions increases the accuracy of intron detection in long-read RNA sequencing.\",\"authors\":\"Matthew T Parker, Katarzyna Knop, Geoffrey J Barton, Gordon G Simpson\",\"doi\":\"10.1186/s13059-021-02296-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Transcription of eukaryotic genomes involves complex alternative processing of RNAs. Sequencing of full-length RNAs using long reads reveals the true complexity of processing. However, the relatively high error rates of long-read sequencing technologies can reduce the accuracy of intron identification. Here we apply alignment metrics and machine-learning-derived sequence information to filter spurious splice junctions from long-read alignments and use the remaining junctions to guide realignment in a two-pass approach. This method, available in the software package 2passtools ( https://github.com/bartongroup/2passtools ), improves the accuracy of spliced alignment and transcriptome assembly for species both with and without existing high-quality annotations.</p>\",\"PeriodicalId\":48922,\"journal\":{\"name\":\"Genome Biology\",\"volume\":\"22 1\",\"pages\":\"72\"},\"PeriodicalIF\":12.3000,\"publicationDate\":\"2021-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13059-021-02296-0\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13059-021-02296-0\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13059-021-02296-0","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
2passtools: two-pass alignment using machine-learning-filtered splice junctions increases the accuracy of intron detection in long-read RNA sequencing.
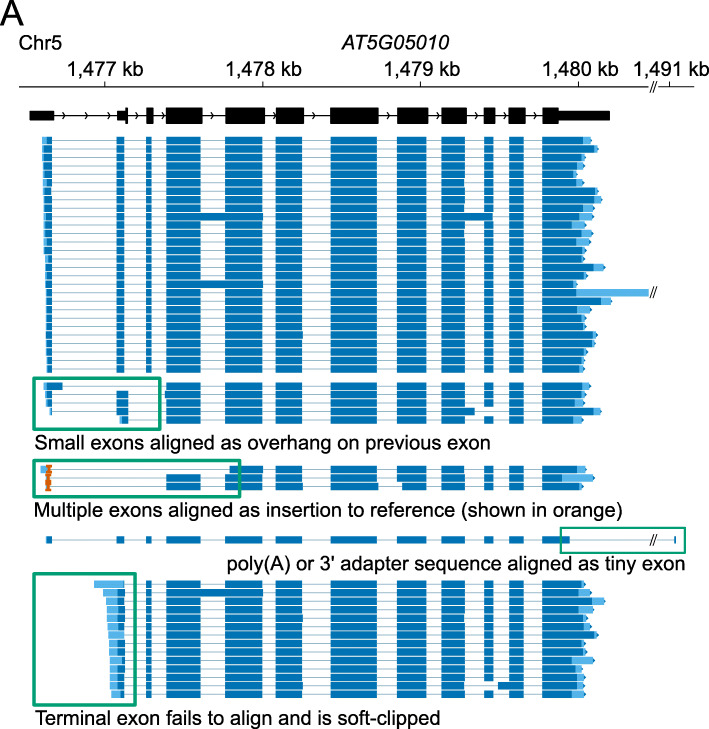
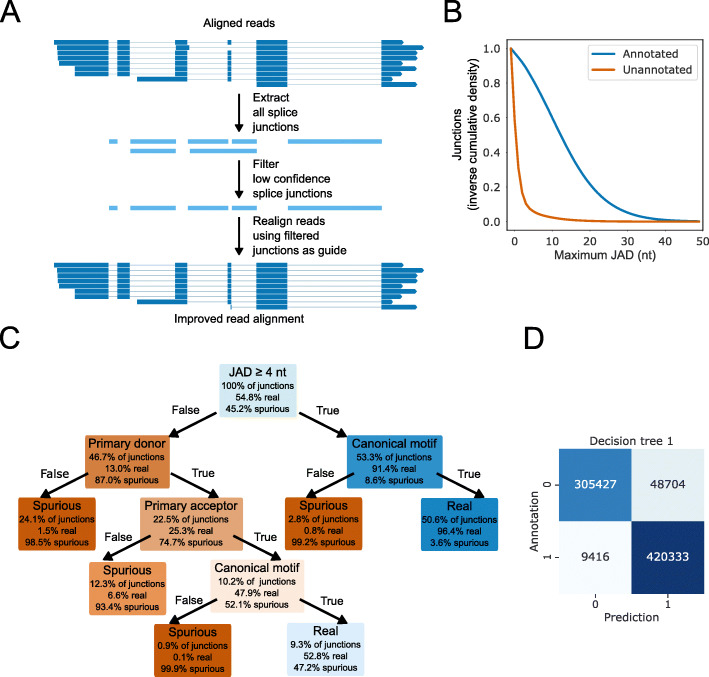
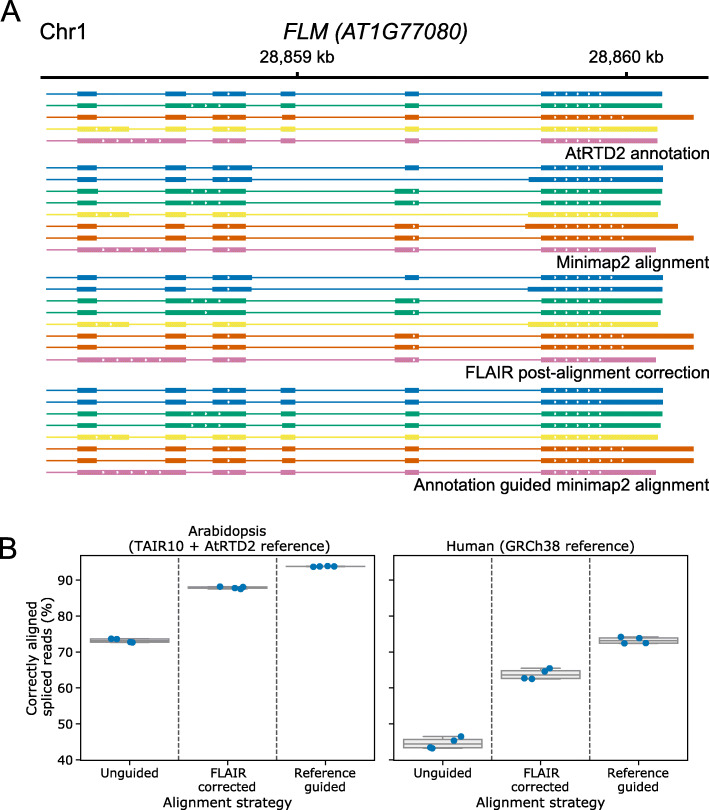
Transcription of eukaryotic genomes involves complex alternative processing of RNAs. Sequencing of full-length RNAs using long reads reveals the true complexity of processing. However, the relatively high error rates of long-read sequencing technologies can reduce the accuracy of intron identification. Here we apply alignment metrics and machine-learning-derived sequence information to filter spurious splice junctions from long-read alignments and use the remaining junctions to guide realignment in a two-pass approach. This method, available in the software package 2passtools ( https://github.com/bartongroup/2passtools ), improves the accuracy of spliced alignment and transcriptome assembly for species both with and without existing high-quality annotations.
期刊介绍:
Genome Biology is a leading research journal that focuses on the study of biology and biomedicine from a genomic and post-genomic standpoint. The journal consistently publishes outstanding research across various areas within these fields.
With an impressive impact factor of 12.3 (2022), Genome Biology has earned its place as the 3rd highest-ranked research journal in the Genetics and Heredity category, according to Thomson Reuters. Additionally, it is ranked 2nd among research journals in the Biotechnology and Applied Microbiology category. It is important to note that Genome Biology is the top-ranking open access journal in this category.
In summary, Genome Biology sets a high standard for scientific publications in the field, showcasing cutting-edge research and earning recognition among its peers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


