Stefan Kohl, Sandra Habbig, Lutz T Weber, Max C Liebau
{"title":"先天性肾和尿路异常(CAKUT)的分子原因。","authors":"Stefan Kohl, Sandra Habbig, Lutz T Weber, Max C Liebau","doi":"10.1186/s40348-021-00112-0","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital anomalies of the kidney and urinary tract (CAKUT) occur in 0.5-1/100 newborns and as a group they represent the most frequent cause for chronic kidney failure in children. CAKUT comprise clinically heterogeneous conditions, ranging from mild vesicoureteral reflux to kidney aplasia. Most forms of CAKUT share the pathophysiology of an impaired developmental interaction of the ureteric bud (UB) and the metanephric mesenchyme (MM). In most cases, CAKUT present as an isolated condition. They also may occur as a component in rare multi-organ syndromes. Many CAKUT probably have a multifactorial etiology. However, up to 20% of human patients and > 200 transgenic mouse models have a monogenic form of CAKUT, which has fueled our efforts to unravel molecular kidney (mal-)development. To date, genetic variants in more than 50 genes have been associated with (isolated) CAKUT in humans. In this short review, we will summarize typical imaging findings in patients with CAKUT and highlight recent mechanistic insight in the molecular pathogenesis of monogenic forms of CAKUT.</p>","PeriodicalId":74215,"journal":{"name":"Molecular and cellular pediatrics","volume":"8 1","pages":"2"},"PeriodicalIF":3.4000,"publicationDate":"2021-02-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40348-021-00112-0","citationCount":"17","resultStr":"{\"title\":\"Molecular causes of congenital anomalies of the kidney and urinary tract (CAKUT).\",\"authors\":\"Stefan Kohl, Sandra Habbig, Lutz T Weber, Max C Liebau\",\"doi\":\"10.1186/s40348-021-00112-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Congenital anomalies of the kidney and urinary tract (CAKUT) occur in 0.5-1/100 newborns and as a group they represent the most frequent cause for chronic kidney failure in children. CAKUT comprise clinically heterogeneous conditions, ranging from mild vesicoureteral reflux to kidney aplasia. Most forms of CAKUT share the pathophysiology of an impaired developmental interaction of the ureteric bud (UB) and the metanephric mesenchyme (MM). In most cases, CAKUT present as an isolated condition. They also may occur as a component in rare multi-organ syndromes. Many CAKUT probably have a multifactorial etiology. However, up to 20% of human patients and > 200 transgenic mouse models have a monogenic form of CAKUT, which has fueled our efforts to unravel molecular kidney (mal-)development. To date, genetic variants in more than 50 genes have been associated with (isolated) CAKUT in humans. In this short review, we will summarize typical imaging findings in patients with CAKUT and highlight recent mechanistic insight in the molecular pathogenesis of monogenic forms of CAKUT.</p>\",\"PeriodicalId\":74215,\"journal\":{\"name\":\"Molecular and cellular pediatrics\",\"volume\":\"8 1\",\"pages\":\"2\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2021-02-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s40348-021-00112-0\",\"citationCount\":\"17\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular and cellular pediatrics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s40348-021-00112-0\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular and cellular pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40348-021-00112-0","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
Molecular causes of congenital anomalies of the kidney and urinary tract (CAKUT).
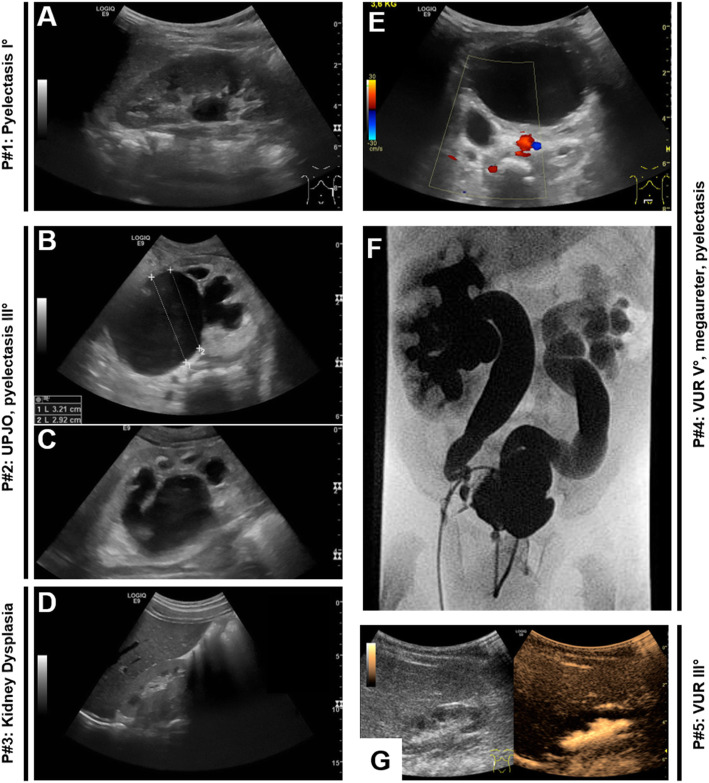
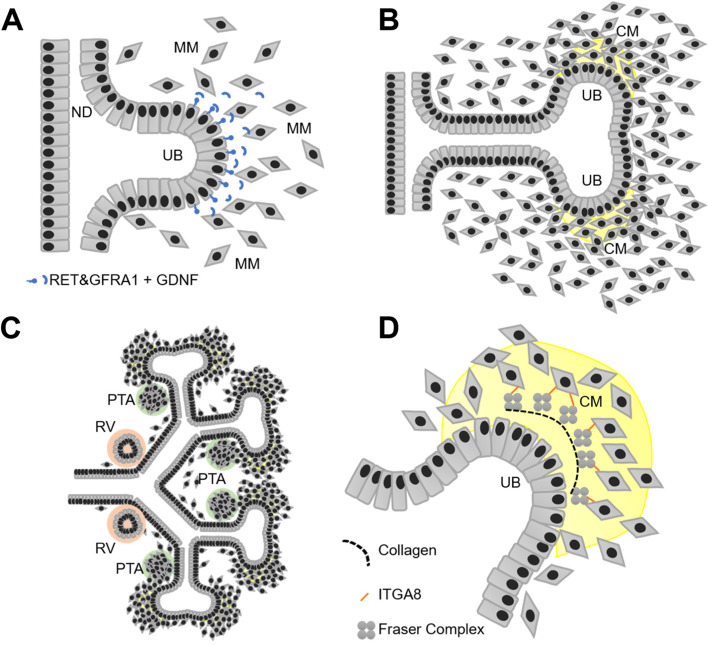
Congenital anomalies of the kidney and urinary tract (CAKUT) occur in 0.5-1/100 newborns and as a group they represent the most frequent cause for chronic kidney failure in children. CAKUT comprise clinically heterogeneous conditions, ranging from mild vesicoureteral reflux to kidney aplasia. Most forms of CAKUT share the pathophysiology of an impaired developmental interaction of the ureteric bud (UB) and the metanephric mesenchyme (MM). In most cases, CAKUT present as an isolated condition. They also may occur as a component in rare multi-organ syndromes. Many CAKUT probably have a multifactorial etiology. However, up to 20% of human patients and > 200 transgenic mouse models have a monogenic form of CAKUT, which has fueled our efforts to unravel molecular kidney (mal-)development. To date, genetic variants in more than 50 genes have been associated with (isolated) CAKUT in humans. In this short review, we will summarize typical imaging findings in patients with CAKUT and highlight recent mechanistic insight in the molecular pathogenesis of monogenic forms of CAKUT.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


