Raí F. Jucá, José Gadelha da Silva Filho, Lindemberg S. Oliveira, Antônio Joel Ramiro de Castro, Marcelo A. S. Silva, Antonio Sérgio B. Sombra, Pierre Basílio Almeida Fechine, João Maria Soares, Antônio César Honorato Barreto, Paulo T. C. Freire, Gilberto Dantas Saraiva
{"title":"β-Bi2Mo2O9 Dibismuth二钼结构、电子和振动特性的实验与理论研究","authors":"Raí F. Jucá, José Gadelha da Silva Filho, Lindemberg S. Oliveira, Antônio Joel Ramiro de Castro, Marcelo A. S. Silva, Antonio Sérgio B. Sombra, Pierre Basílio Almeida Fechine, João Maria Soares, Antônio César Honorato Barreto, Paulo T. C. Freire, Gilberto Dantas Saraiva","doi":"10.1021/acs.jpcc.5c04920","DOIUrl":null,"url":null,"abstract":"This study presents a comprehensive structural, vibrational, and electronic investigation of the monoclinic β-Bi<sub>2</sub>Mo<sub>2</sub>O<sub>9</sub> compound, employing experimental and first-principles approaches. X-ray diffraction combined with Rietveld refinement confirms the crystallization of Bi<sub>2</sub>Mo<sub>2</sub>O<sub>9</sub> in the P2<sub>1</sub>/n space group. Density functional theory calculations within the LDA-D framework reveal a slight underestimation of the lattice parameters and unit cell volume, while preserving local geometries. The vibrational properties were examined through Raman and infrared spectroscopy, supported by group-theory analysis, indicating a rich phonon activity consistent with the complex symmetry and multiatom basis of the monoclinic lattice. Electronic structure calculations identify BMO as an indirect band gap semiconductor with a computed band gap of 2.23 eV, closely matching experimental optical data. Bader charge and electron localization function (ELF) analyses highlight a mixed ionic–covalent bonding character, particularly pronounced in the Mo–O sublattice. Low-temperature Raman spectra (12–300 K) revealed systematic redshifts, peak broadenings, and intensity reductions across low- and high-wavenumber phonon modes, indicative of pronounced anharmonic effects and thermal expansion of the lattice. On the contrary, high-pressure Raman spectra collected up to 9.08 GPa showed a general phonon hardening trend with increasing pressure, attributable to lattice compression. Several Raman modes exhibited anomalous behavior, such as mode appearance, disappearance, and slope changes, pointing to pressure-induced phase transitions and potential symmetry changes.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"58 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2025-10-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Experimental and Theoretical Investigations on the Structural, Electronic, and Vibrational Properties of β-Bi2Mo2O9 Dibismuth Dimolybdenum\",\"authors\":\"Raí F. Jucá, José Gadelha da Silva Filho, Lindemberg S. Oliveira, Antônio Joel Ramiro de Castro, Marcelo A. S. Silva, Antonio Sérgio B. Sombra, Pierre Basílio Almeida Fechine, João Maria Soares, Antônio César Honorato Barreto, Paulo T. C. Freire, Gilberto Dantas Saraiva\",\"doi\":\"10.1021/acs.jpcc.5c04920\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"This study presents a comprehensive structural, vibrational, and electronic investigation of the monoclinic β-Bi<sub>2</sub>Mo<sub>2</sub>O<sub>9</sub> compound, employing experimental and first-principles approaches. X-ray diffraction combined with Rietveld refinement confirms the crystallization of Bi<sub>2</sub>Mo<sub>2</sub>O<sub>9</sub> in the P2<sub>1</sub>/n space group. Density functional theory calculations within the LDA-D framework reveal a slight underestimation of the lattice parameters and unit cell volume, while preserving local geometries. The vibrational properties were examined through Raman and infrared spectroscopy, supported by group-theory analysis, indicating a rich phonon activity consistent with the complex symmetry and multiatom basis of the monoclinic lattice. Electronic structure calculations identify BMO as an indirect band gap semiconductor with a computed band gap of 2.23 eV, closely matching experimental optical data. Bader charge and electron localization function (ELF) analyses highlight a mixed ionic–covalent bonding character, particularly pronounced in the Mo–O sublattice. Low-temperature Raman spectra (12–300 K) revealed systematic redshifts, peak broadenings, and intensity reductions across low- and high-wavenumber phonon modes, indicative of pronounced anharmonic effects and thermal expansion of the lattice. On the contrary, high-pressure Raman spectra collected up to 9.08 GPa showed a general phonon hardening trend with increasing pressure, attributable to lattice compression. Several Raman modes exhibited anomalous behavior, such as mode appearance, disappearance, and slope changes, pointing to pressure-induced phase transitions and potential symmetry changes.\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"58 1\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-10-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpcc.5c04920\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.5c04920","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Experimental and Theoretical Investigations on the Structural, Electronic, and Vibrational Properties of β-Bi2Mo2O9 Dibismuth Dimolybdenum
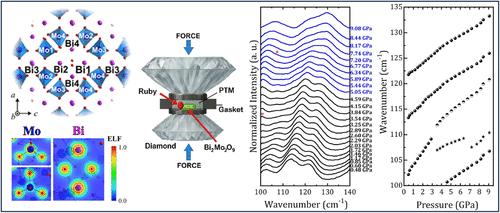
This study presents a comprehensive structural, vibrational, and electronic investigation of the monoclinic β-Bi2Mo2O9 compound, employing experimental and first-principles approaches. X-ray diffraction combined with Rietveld refinement confirms the crystallization of Bi2Mo2O9 in the P21/n space group. Density functional theory calculations within the LDA-D framework reveal a slight underestimation of the lattice parameters and unit cell volume, while preserving local geometries. The vibrational properties were examined through Raman and infrared spectroscopy, supported by group-theory analysis, indicating a rich phonon activity consistent with the complex symmetry and multiatom basis of the monoclinic lattice. Electronic structure calculations identify BMO as an indirect band gap semiconductor with a computed band gap of 2.23 eV, closely matching experimental optical data. Bader charge and electron localization function (ELF) analyses highlight a mixed ionic–covalent bonding character, particularly pronounced in the Mo–O sublattice. Low-temperature Raman spectra (12–300 K) revealed systematic redshifts, peak broadenings, and intensity reductions across low- and high-wavenumber phonon modes, indicative of pronounced anharmonic effects and thermal expansion of the lattice. On the contrary, high-pressure Raman spectra collected up to 9.08 GPa showed a general phonon hardening trend with increasing pressure, attributable to lattice compression. Several Raman modes exhibited anomalous behavior, such as mode appearance, disappearance, and slope changes, pointing to pressure-induced phase transitions and potential symmetry changes.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


