{"title":"基于dft的共价有机框架在吸附、光电、清洁能源存储和气体传感器应用中的评价。","authors":"Abhay P. Srivastava, Brijesh K. Pandey","doi":"10.1007/s00894-025-06535-0","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Covalent Organic Frameworks (COFs), which are frameworks composed of light atoms held together by strong covalent bonds, are generating interest as potential materials for applications such as renewable energy and gas capture. We employed Density Functional Theory (DFT) calculations, as implemented in the VASP code, to look at both 2D and 3D COFs. We systematically analyzed various properties, including structural stability, phonon dispersion, electronic structures, density of states, adsorption behavior, and mechanical properties. To get better accuracy, we took into account van der Waals interactions and even used hybrid functionals. What we found was that 3D COFs generally exhibit greater mechanical strength and, in most cases, better gas adsorption, which seems to come from their interconnected pore structures. On the other hand, 2D COFs exhibit enhanced π-electron delocalization and direct band gaps of approximately 2.5 eV, which may be helpful in sensors and optoelectronics. Phonon analyses verified the dynamical stability of both structures. Ultimately, these results underscore the importance of dimensionality in tailoring COF properties for energy and electronic applications.</p><h3>Method</h3><p>First-principles simulations were performed using Density Functional Theory (DFT) within the Vienna Ab initio Simulation Package (VASP). To account for exchange–correlation effects, we employed the Generalised Gradient Approximation (GGA) in the Perdew–Burke–Ernzerhof (PBE) formulation, and we also utilised projector-augmented wave (PAW) pseudopotentials. Hybrid functional (HSE06) and DFT-D3 van der Waals corrections were introduced to improve the accuracy of our band gap prediction. The plane-wave cutoffs were set at 500 eV for the calculations, and Monkhorst–Pack k-point meshes were used with a 3 × 3 × 1 (2D) and 2 × 2 × 2 (3D) grid. Evaluated were structural optimisations, band structures, total and projected DOS, adsorption energies, and charge transfer (using Bader analysis). Assessment of bonding features utilised the Electron Localisation Function (ELF) and charge density difference (Δρ) visualisations. The Phonopy package was used to calculate Phonon dispersions and thus confirm the dynamic stability of the COFs.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 11","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-10-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"DFT-based evaluation of covalent organic frameworks for adsorption, optoelectronic, clean energy storage, and gas sensor applications\",\"authors\":\"Abhay P. Srivastava, Brijesh K. Pandey\",\"doi\":\"10.1007/s00894-025-06535-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>Covalent Organic Frameworks (COFs), which are frameworks composed of light atoms held together by strong covalent bonds, are generating interest as potential materials for applications such as renewable energy and gas capture. We employed Density Functional Theory (DFT) calculations, as implemented in the VASP code, to look at both 2D and 3D COFs. We systematically analyzed various properties, including structural stability, phonon dispersion, electronic structures, density of states, adsorption behavior, and mechanical properties. To get better accuracy, we took into account van der Waals interactions and even used hybrid functionals. What we found was that 3D COFs generally exhibit greater mechanical strength and, in most cases, better gas adsorption, which seems to come from their interconnected pore structures. On the other hand, 2D COFs exhibit enhanced π-electron delocalization and direct band gaps of approximately 2.5 eV, which may be helpful in sensors and optoelectronics. Phonon analyses verified the dynamical stability of both structures. Ultimately, these results underscore the importance of dimensionality in tailoring COF properties for energy and electronic applications.</p><h3>Method</h3><p>First-principles simulations were performed using Density Functional Theory (DFT) within the Vienna Ab initio Simulation Package (VASP). To account for exchange–correlation effects, we employed the Generalised Gradient Approximation (GGA) in the Perdew–Burke–Ernzerhof (PBE) formulation, and we also utilised projector-augmented wave (PAW) pseudopotentials. Hybrid functional (HSE06) and DFT-D3 van der Waals corrections were introduced to improve the accuracy of our band gap prediction. The plane-wave cutoffs were set at 500 eV for the calculations, and Monkhorst–Pack k-point meshes were used with a 3 × 3 × 1 (2D) and 2 × 2 × 2 (3D) grid. Evaluated were structural optimisations, band structures, total and projected DOS, adsorption energies, and charge transfer (using Bader analysis). Assessment of bonding features utilised the Electron Localisation Function (ELF) and charge density difference (Δρ) visualisations. The Phonopy package was used to calculate Phonon dispersions and thus confirm the dynamic stability of the COFs.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"31 11\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-10-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-025-06535-0\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06535-0","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
DFT-based evaluation of covalent organic frameworks for adsorption, optoelectronic, clean energy storage, and gas sensor applications
Context
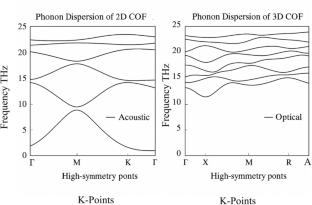
Covalent Organic Frameworks (COFs), which are frameworks composed of light atoms held together by strong covalent bonds, are generating interest as potential materials for applications such as renewable energy and gas capture. We employed Density Functional Theory (DFT) calculations, as implemented in the VASP code, to look at both 2D and 3D COFs. We systematically analyzed various properties, including structural stability, phonon dispersion, electronic structures, density of states, adsorption behavior, and mechanical properties. To get better accuracy, we took into account van der Waals interactions and even used hybrid functionals. What we found was that 3D COFs generally exhibit greater mechanical strength and, in most cases, better gas adsorption, which seems to come from their interconnected pore structures. On the other hand, 2D COFs exhibit enhanced π-electron delocalization and direct band gaps of approximately 2.5 eV, which may be helpful in sensors and optoelectronics. Phonon analyses verified the dynamical stability of both structures. Ultimately, these results underscore the importance of dimensionality in tailoring COF properties for energy and electronic applications.
Method
First-principles simulations were performed using Density Functional Theory (DFT) within the Vienna Ab initio Simulation Package (VASP). To account for exchange–correlation effects, we employed the Generalised Gradient Approximation (GGA) in the Perdew–Burke–Ernzerhof (PBE) formulation, and we also utilised projector-augmented wave (PAW) pseudopotentials. Hybrid functional (HSE06) and DFT-D3 van der Waals corrections were introduced to improve the accuracy of our band gap prediction. The plane-wave cutoffs were set at 500 eV for the calculations, and Monkhorst–Pack k-point meshes were used with a 3 × 3 × 1 (2D) and 2 × 2 × 2 (3D) grid. Evaluated were structural optimisations, band structures, total and projected DOS, adsorption energies, and charge transfer (using Bader analysis). Assessment of bonding features utilised the Electron Localisation Function (ELF) and charge density difference (Δρ) visualisations. The Phonopy package was used to calculate Phonon dispersions and thus confirm the dynamic stability of the COFs.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


