{"title":"SAGPEK:快速和灵活的方法来确定基因型的Sanger测序数据。","authors":"Jinpeng Wang, Shuo Sun, Yaran Zhang, Ning Huang, Chunhong Yang, Yaping Gao, Xiuge Wang, Zhihua Ju, Qiang Jiang, Yao Xiao, Xiaochao Wei, Wenhao Liu, Jinming Huang","doi":"10.1186/s12859-025-06271-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Although Sanger sequencing remains widely used in human genetic disease diagnosis and livestock breeding, software packages for analyzing such data have seen little innovation over time. Determining the genotypes of tens to hundreds of loci across hundreds or thousands of samples still typically relies on manual visual confirmation with traditional software, a process that is both time-consuming and prone to error.</p><p><strong>Results: </strong>We present SAGPEK, a tool that automatically identifies genotypes at target loci from hundreds to thousands of ABI-format Sanger sequencing files and directly outputs the results. SAGPEK extracts the signal intensities for A, G, C, and T bases, performs base calling, and determines each site's homozygous or heterozygous status. It then generates a primary sequence composed of the bases with the highest signal intensities and records secondary bases for heterozygous sites. Using either built-in or user-provided anchor sequences, SAGPEK maps the coordinates of target loci, reports their genotypes, and, when applicable, annotates the corresponding amino acid changes.</p><p><strong>Conclusions: </strong>SAGPEK provides an efficient, flexible, and user-friendly solution for analyzing ABI-format Sanger sequencing data, enabling simultaneous genotyping of tens of loci across hundreds of samples. Its innovation lies not in introducing new base-calling methods, but in integrating versatile functionalities-batch genotyping, customizable anchor sequences, amino acid alteration reporting, chromatogram visualization, and local execution-into a single open-source package. This makes SAGPEK well suited for applications such as human genetic disease screening, drug-resistance mutation detection, and functional mutation identification in livestock and other organisms.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"246"},"PeriodicalIF":3.3000,"publicationDate":"2025-10-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12523141/pdf/","citationCount":"0","resultStr":"{\"title\":\"SAGPEK: fast and flexible approach to identify genotypes of Sanger sequencing data.\",\"authors\":\"Jinpeng Wang, Shuo Sun, Yaran Zhang, Ning Huang, Chunhong Yang, Yaping Gao, Xiuge Wang, Zhihua Ju, Qiang Jiang, Yao Xiao, Xiaochao Wei, Wenhao Liu, Jinming Huang\",\"doi\":\"10.1186/s12859-025-06271-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Although Sanger sequencing remains widely used in human genetic disease diagnosis and livestock breeding, software packages for analyzing such data have seen little innovation over time. Determining the genotypes of tens to hundreds of loci across hundreds or thousands of samples still typically relies on manual visual confirmation with traditional software, a process that is both time-consuming and prone to error.</p><p><strong>Results: </strong>We present SAGPEK, a tool that automatically identifies genotypes at target loci from hundreds to thousands of ABI-format Sanger sequencing files and directly outputs the results. SAGPEK extracts the signal intensities for A, G, C, and T bases, performs base calling, and determines each site's homozygous or heterozygous status. It then generates a primary sequence composed of the bases with the highest signal intensities and records secondary bases for heterozygous sites. Using either built-in or user-provided anchor sequences, SAGPEK maps the coordinates of target loci, reports their genotypes, and, when applicable, annotates the corresponding amino acid changes.</p><p><strong>Conclusions: </strong>SAGPEK provides an efficient, flexible, and user-friendly solution for analyzing ABI-format Sanger sequencing data, enabling simultaneous genotyping of tens of loci across hundreds of samples. Its innovation lies not in introducing new base-calling methods, but in integrating versatile functionalities-batch genotyping, customizable anchor sequences, amino acid alteration reporting, chromatogram visualization, and local execution-into a single open-source package. This makes SAGPEK well suited for applications such as human genetic disease screening, drug-resistance mutation detection, and functional mutation identification in livestock and other organisms.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"26 1\",\"pages\":\"246\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-10-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12523141/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-025-06271-5\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06271-5","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
SAGPEK: fast and flexible approach to identify genotypes of Sanger sequencing data.
Background: Although Sanger sequencing remains widely used in human genetic disease diagnosis and livestock breeding, software packages for analyzing such data have seen little innovation over time. Determining the genotypes of tens to hundreds of loci across hundreds or thousands of samples still typically relies on manual visual confirmation with traditional software, a process that is both time-consuming and prone to error.
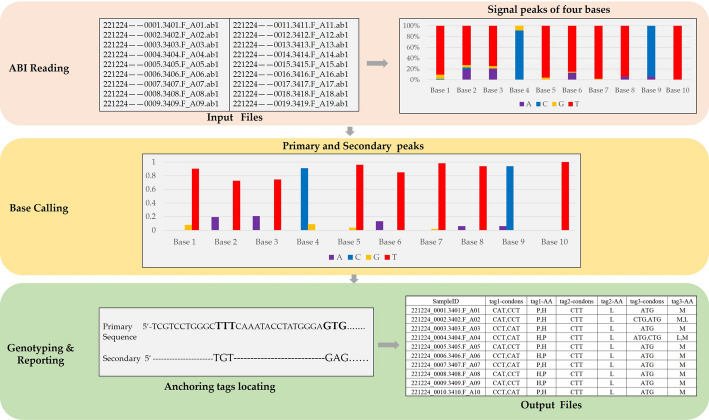
Results: We present SAGPEK, a tool that automatically identifies genotypes at target loci from hundreds to thousands of ABI-format Sanger sequencing files and directly outputs the results. SAGPEK extracts the signal intensities for A, G, C, and T bases, performs base calling, and determines each site's homozygous or heterozygous status. It then generates a primary sequence composed of the bases with the highest signal intensities and records secondary bases for heterozygous sites. Using either built-in or user-provided anchor sequences, SAGPEK maps the coordinates of target loci, reports their genotypes, and, when applicable, annotates the corresponding amino acid changes.
Conclusions: SAGPEK provides an efficient, flexible, and user-friendly solution for analyzing ABI-format Sanger sequencing data, enabling simultaneous genotyping of tens of loci across hundreds of samples. Its innovation lies not in introducing new base-calling methods, but in integrating versatile functionalities-batch genotyping, customizable anchor sequences, amino acid alteration reporting, chromatogram visualization, and local execution-into a single open-source package. This makes SAGPEK well suited for applications such as human genetic disease screening, drug-resistance mutation detection, and functional mutation identification in livestock and other organisms.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


