利用动态替代模型在铁基双金属表面上直接脱氧苯酚
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
我们使用最近开发的高斯过程回归(GPR)计算器对铁基双金属表面上的苯酚直接脱氧(DDO)进行了加速轻推弹性带(NEB)研究。我们的测试计算表明,与传统密度泛函理论计算相比,GPR计算器在保持高精度的同时实现了高达3倍的加速,能量势垒误差低于0.015 eV。利用GPR-NEB,我们系统地研究了纯Fe(110)和表层和次表层Co和Ni修饰的表面的DDO机制。我们的研究结果表明,与纯Fe(110)表面相比,Co和Ni的亚表面取代保持了良好的C-O键裂解和C-H键形成的热力学和动力学。相比之下,顶层取代通常会增加C-O键的解理障,使步骤吸热,并导致明显更高的逆反应速率,使得DDO在这些表面上不利。这项工作证明了grr加速过渡态搜索复杂表面反应的有效性,并为选择性脱氧双金属催化剂的合理设计提供了见解。本文章由计算机程序翻译,如有差异,请以英文原文为准。
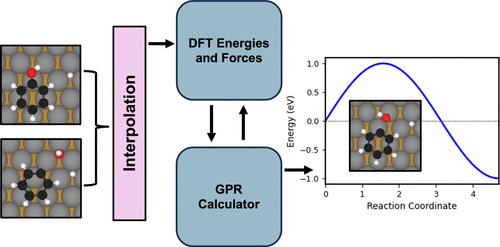
Direct Deoxygenation of Phenol over Fe-Based Bimetallic Surfaces Using On-the-Fly Surrogate Models
We present an accelerated nudged elastic band (NEB) study of phenol direct deoxygenation (DDO) on Fe-based bimetallic surfaces using a recently developed Gaussian process regression (GPR) calculator. Our test calculations demonstrate that the GPR calculator achieves up to 3 times speedup compared to conventional density functional theory calculations while maintaining high accuracy, with energy barrier errors below 0.015 eV. Using GPR-NEB, we systematically examine the DDO mechanism on pure Fe(110) and surfaces modified with Co and Ni in both top and subsurface layers. Our results show that subsurface Co and Ni substitutions preserve favorable thermodynamics and kinetics for both C–O bond cleavage and C–H bond formation, comparable to those on the pure Fe(110) surface. In contrast, top-layer substitutions generally increase the C–O bond cleavage barrier, render the step endothermic, and result in significantly higher reverse reaction rates, making DDO unfavorable on these surfaces. This work demonstrates the effectiveness of GRR-accelerated transition state searches for complex surface reactions and provides insights into rational design of bimetallic catalysts for selective deoxygenation.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


