Aaron M Walsh, Emma Roycroft, Kate Hinchion, Sharee A Basdeo, Frederick J Sheedy, Fiona Crispie, Paul D Cotter, Anne Marie McLaughlin, Joseph Keane, Margaret M Fitzgibbon, Laura E Gleeson
{"title":"来自慢性感染患者的复发性禽分枝杆菌分离株的基因组特征揭示了宿主内进化的模式。","authors":"Aaron M Walsh, Emma Roycroft, Kate Hinchion, Sharee A Basdeo, Frederick J Sheedy, Fiona Crispie, Paul D Cotter, Anne Marie McLaughlin, Joseph Keane, Margaret M Fitzgibbon, Laura E Gleeson","doi":"10.1186/s13073-025-01549-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Mycobacterium avium complex causes chronic and difficult-to-treat infection in vulnerable patient groups, and incidence is increasing worldwide. Whole genome sequencing has the potential to reveal new information about how M. avium persists over time in the human lung.</p><p><strong>Methods: </strong>We analysed the genomes of 287 isolates of M. avium that were sampled longitudinally from 56 patients. Our dataset included 50 newly sequenced genomes from a cohort of 20 patients from Ireland who were sampled for up to 10 years, and we compared these to 237 published genomes from 2 pre-existing cohorts from Europe to evaluate strains from Ireland in a wider context. Additionally, we performed a combined analysis across the 3 cohorts to examine the changes that occurred over the course of infection.</p><p><strong>Results: </strong>We identified 2 instances where strains from Ireland clustered with strains from Europe within a 13-SNP threshold, supporting previous observations that dominant circulating clones of M. avium are present internationally. Across the 3 cohorts, we found that the communities of M. avium evolved over time within individual hosts, and we report that acquisition of new strains is frequent. Importantly, our findings suggest that M. avium may adapt to the conditions that it faces in the host, with evidence of positive selection of 13 distinct mycobacterial genes. Notably, multiple virulence-associated genes were under selection, including genes that could confer resistance to antibiotics and host defence mechanisms.</p><p><strong>Conclusions: </strong>Whole genome sequencing provides novel insights into within-host evolution of M. avium and highlights potentially important mycobacterial strategies to enhance persistence that may provide new targets for therapeutic investigation.</p>","PeriodicalId":12645,"journal":{"name":"Genome Medicine","volume":"17 1","pages":"120"},"PeriodicalIF":10.4000,"publicationDate":"2025-10-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12516842/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genomic characterisation of recurrent Mycobacterium avium isolates from chronically infected patients reveals patterns of within-host evolution.\",\"authors\":\"Aaron M Walsh, Emma Roycroft, Kate Hinchion, Sharee A Basdeo, Frederick J Sheedy, Fiona Crispie, Paul D Cotter, Anne Marie McLaughlin, Joseph Keane, Margaret M Fitzgibbon, Laura E Gleeson\",\"doi\":\"10.1186/s13073-025-01549-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Mycobacterium avium complex causes chronic and difficult-to-treat infection in vulnerable patient groups, and incidence is increasing worldwide. Whole genome sequencing has the potential to reveal new information about how M. avium persists over time in the human lung.</p><p><strong>Methods: </strong>We analysed the genomes of 287 isolates of M. avium that were sampled longitudinally from 56 patients. Our dataset included 50 newly sequenced genomes from a cohort of 20 patients from Ireland who were sampled for up to 10 years, and we compared these to 237 published genomes from 2 pre-existing cohorts from Europe to evaluate strains from Ireland in a wider context. Additionally, we performed a combined analysis across the 3 cohorts to examine the changes that occurred over the course of infection.</p><p><strong>Results: </strong>We identified 2 instances where strains from Ireland clustered with strains from Europe within a 13-SNP threshold, supporting previous observations that dominant circulating clones of M. avium are present internationally. Across the 3 cohorts, we found that the communities of M. avium evolved over time within individual hosts, and we report that acquisition of new strains is frequent. Importantly, our findings suggest that M. avium may adapt to the conditions that it faces in the host, with evidence of positive selection of 13 distinct mycobacterial genes. Notably, multiple virulence-associated genes were under selection, including genes that could confer resistance to antibiotics and host defence mechanisms.</p><p><strong>Conclusions: </strong>Whole genome sequencing provides novel insights into within-host evolution of M. avium and highlights potentially important mycobacterial strategies to enhance persistence that may provide new targets for therapeutic investigation.</p>\",\"PeriodicalId\":12645,\"journal\":{\"name\":\"Genome Medicine\",\"volume\":\"17 1\",\"pages\":\"120\"},\"PeriodicalIF\":10.4000,\"publicationDate\":\"2025-10-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12516842/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Medicine\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13073-025-01549-y\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Medicine","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13073-025-01549-y","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genomic characterisation of recurrent Mycobacterium avium isolates from chronically infected patients reveals patterns of within-host evolution.
Background: Mycobacterium avium complex causes chronic and difficult-to-treat infection in vulnerable patient groups, and incidence is increasing worldwide. Whole genome sequencing has the potential to reveal new information about how M. avium persists over time in the human lung.
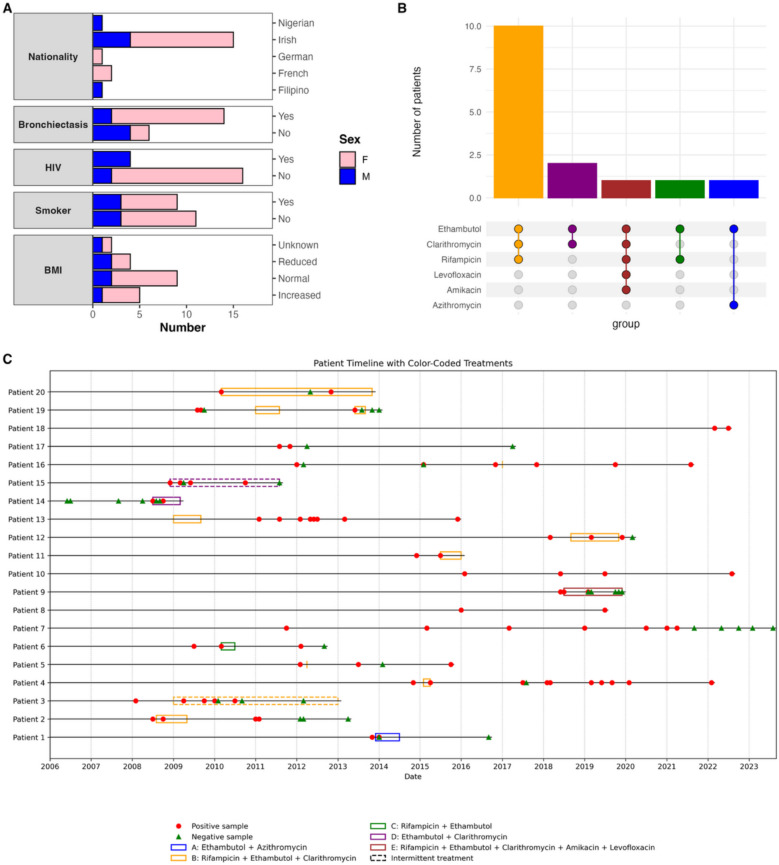
Methods: We analysed the genomes of 287 isolates of M. avium that were sampled longitudinally from 56 patients. Our dataset included 50 newly sequenced genomes from a cohort of 20 patients from Ireland who were sampled for up to 10 years, and we compared these to 237 published genomes from 2 pre-existing cohorts from Europe to evaluate strains from Ireland in a wider context. Additionally, we performed a combined analysis across the 3 cohorts to examine the changes that occurred over the course of infection.
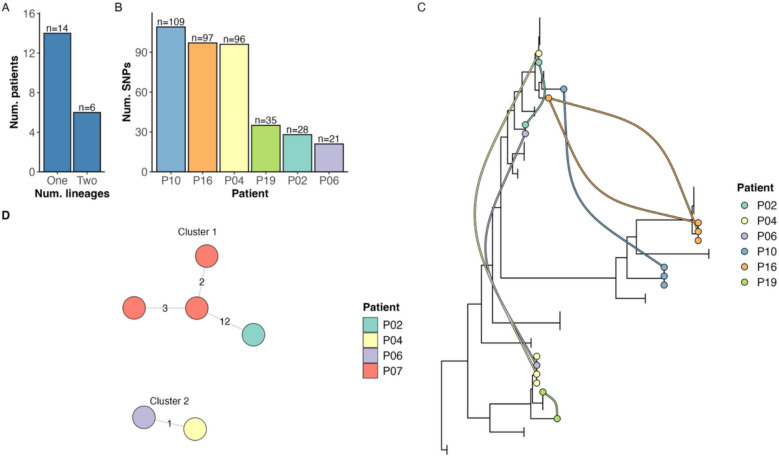
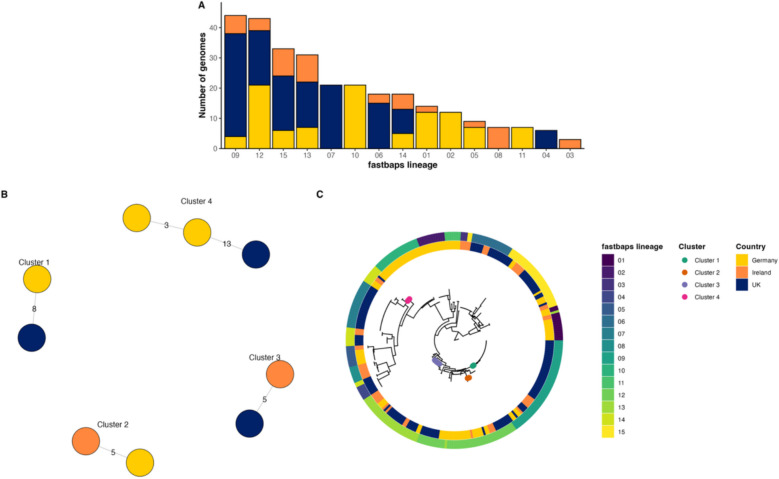
Results: We identified 2 instances where strains from Ireland clustered with strains from Europe within a 13-SNP threshold, supporting previous observations that dominant circulating clones of M. avium are present internationally. Across the 3 cohorts, we found that the communities of M. avium evolved over time within individual hosts, and we report that acquisition of new strains is frequent. Importantly, our findings suggest that M. avium may adapt to the conditions that it faces in the host, with evidence of positive selection of 13 distinct mycobacterial genes. Notably, multiple virulence-associated genes were under selection, including genes that could confer resistance to antibiotics and host defence mechanisms.
Conclusions: Whole genome sequencing provides novel insights into within-host evolution of M. avium and highlights potentially important mycobacterial strategies to enhance persistence that may provide new targets for therapeutic investigation.
期刊介绍:
Genome Medicine is an open access journal that publishes outstanding research applying genetics, genomics, and multi-omics to understand, diagnose, and treat disease. Bridging basic science and clinical research, it covers areas such as cancer genomics, immuno-oncology, immunogenomics, infectious disease, microbiome, neurogenomics, systems medicine, clinical genomics, gene therapies, precision medicine, and clinical trials. The journal publishes original research, methods, software, and reviews to serve authors and promote broad interest and importance in the field.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


