二维单硫系材料设计的桥接文本挖掘和量子模拟
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
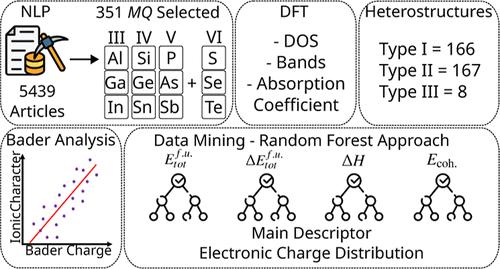
建立一个结构化的框架,致力于系统地回顾文献、识别潜在化合物、理论量子化学表征和评估稳定性描述符的重要性,对于加速发现用于技术应用的二维材料至关重要。在这项工作中,我们选择了二维单硫族化合物(MQ)作为本研究的原型材料,由于它们在未来设备中的潜在应用而引起了越来越多的兴趣。我们的框架从对5400多篇文章的自然语言处理分析开始,揭示了对二维MQ化合物日益增长的研究兴趣,特别是在电子、能源和基础研究方面。这导致在13种不同的二维结构相中选择27种不同的MQ化合物进行密度泛函理论计算,从而形成了一个广泛的物理化学性质数据库。我们评估了生成焓,发现了明确的稳定性趋势(例如,随着较重的硫,稳定性下降,GeSe的动态鲁棒性),并评估了平衡晶格参数,注意到可预测的膨胀和显著的各向异性。Bader电荷分析提供了对电荷转移和离子性趋势的见解。我们的电子结构分析发现带隙(直接或间接,取决于M元素族和硫的重量)和光学吸收各向异性受到晶体对称性和自旋轨道耦合的显著影响。重要的是,带对准计算将所有可能的异质结分类为I型,II型或III型,强调了它们在未来光电器件发展中的广泛潜力。此外,我们结合了机器学习,使用随机森林方法,以及我们的密度泛函理论计算来准确预测能量特性的趋势。该分析将电子电荷确定为34个描述符中非常重要的稳定性描述符,强调了其对未来机器学习努力的重要性。因此,本研究为有前途的二维材料的表征和发现提供了一个简化的框架,突出了数据驱动分析和基于量子的模拟之间的协同作用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Bridging Text Mining and Quantum Simulations for the Design of 2D Monochalcogenide Materials
The formulation of a structured framework dedicated to the systematic review of the literature, identification of potential compounds, theoretical quantum chemistry characterization, and assessment of the significance of stability descriptors is essential for accelerating the discovery of two-dimensional materials for technological applications. In this work, we selected the two-dimensional monochalcogenides (MQ), which have attracted increased interest due to their potential applications in future devices, as prototype materials for the present investigation. Our framework started with a natural language processing analysis of more than 5400 articles, revealing a growing research interest in the two-dimensional MQ compounds, especially in electronics, energy, and fundamental studies. This led to the selection of 27 diverse MQ compounds across 13 distinct two-dimensional structural phases for density functional theory calculations, resulting in an extensive database of physicochemical properties. We evaluated formation enthalpies, uncovering clear stability trends (e.g., stability declines with heavier chalcogens, dynamic robustness of GeSe), and evaluated equilibrium lattice parameters, noting predictable expansions and significant anisotropies. Bader charge analysis offered insights into charge-transfer and ionicity trends. Our electronic structure analysis identified band gaps (direct or indirect, depending on the M element group and the weight of the chalcogen) and optical absorption anisotropies significantly influenced by crystal symmetry and spin orbit coupling. Importantly, band alignment calculations classified all possible heterojunctions as type I, II, or III, underscoring their extensive potential for future optoelectronic device development. Additionally, we incorporated machine learning, using a random forest approach, along with our density functional theory calculations to accurately forecast trends in energetic properties. This analysis identified the electronic charge as a highly significant stability descriptor among the 34 descriptors, underscoring its importance for future machine learning endeavors. Hence, this study offers a streamlined framework for the characterization and discovery of promising two-dimensional materials, highlighting the synergy between data-driven analysis and quantum-based simulations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


