{"title":"通过单细胞RNA测序和机器学习方法开发和验证白血病预后模型。","authors":"Doujia Chen, Jie Yang, Mengting Wang, Tianye Jian","doi":"10.1007/s12672-025-03757-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Leukemia prognosis varies significantly among patients, highlighting the need for accurate prediction tools. Emerging evidence suggests that the immune microenvironment plays a crucial role in leukemia progression and treatment response.</p><p><strong>Methods: </strong>We analyzed RNA expression profiles and clinical data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases, supplemented by single-cell RNA sequencing datasets. Differential gene expression analysis was performed using stringent criteria (logFC > 1, FDR < 0.05) to identify leukemia-associated genes. Ten distinct machine learning algorithms, including Lasso, CoxBoost, and ensemble methods, were implemented for prognostic model development with cross-platform validation. Single-cell analysis employed Seurat for quality control and cell type annotation, while CellChat algorithm mapped intercellular communication networks. Experimental validation was conducted using quantitative RT-PCR analysis of key immune markers (TLR2, TLR4, CCR7, IL18) in U937 and K562 leukemia cell lines compared to normal peripheral blood mononuclear cells.</p><p><strong>Results: </strong>The machine learning-derived prognostic model demonstrated exceptional predictive performance with area under the curve values of 0.874, 0.891, and 0.925 for 1-, 2-, and 3-year survival endpoints, respectively. Six critical immune regulatory genes (TLR2, TLR4, CCR7, IL18, TIRAP, FOXP3) were identified as both differentially expressed and prognostically significant, with IL18 showing the highest discriminative capacity (AUC = 0.983). RT-PCR validation confirmed significant upregulation of all tested genes in leukemia cell lines: TLR2 (3.8-fold in U937, 2.2-fold in K562), TLR4 (3.4-fold in U937, 1.8-fold in K562), CCR7 (4.1-fold in U937, 2.7-fold in K562), and IL18 (5.2-fold in U937, 3.6-fold in K562) compared to normal controls (all p < 0.05). Single-cell analysis revealed substantial cellular heterogeneity with cell type-specific expression patterns and complex intercellular communication networks involving B cells, T cells, natural killer cells, and dendritic cells.</p><p><strong>Conclusion: </strong>This study provides a reliable prognostic tool for leukemia and offers insights into the critical role of the immune microenvironment in leukemia pathogenesis. Our findings may guide the development of personalized immunotherapy strategies for leukemia patients.</p>","PeriodicalId":11148,"journal":{"name":"Discover. Oncology","volume":"16 1","pages":"1846"},"PeriodicalIF":2.9000,"publicationDate":"2025-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12514105/pdf/","citationCount":"0","resultStr":"{\"title\":\"Development and validation of a leukemia prognostic model through single-cell RNA sequencing and machine learning approaches.\",\"authors\":\"Doujia Chen, Jie Yang, Mengting Wang, Tianye Jian\",\"doi\":\"10.1007/s12672-025-03757-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Leukemia prognosis varies significantly among patients, highlighting the need for accurate prediction tools. Emerging evidence suggests that the immune microenvironment plays a crucial role in leukemia progression and treatment response.</p><p><strong>Methods: </strong>We analyzed RNA expression profiles and clinical data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases, supplemented by single-cell RNA sequencing datasets. Differential gene expression analysis was performed using stringent criteria (logFC > 1, FDR < 0.05) to identify leukemia-associated genes. Ten distinct machine learning algorithms, including Lasso, CoxBoost, and ensemble methods, were implemented for prognostic model development with cross-platform validation. Single-cell analysis employed Seurat for quality control and cell type annotation, while CellChat algorithm mapped intercellular communication networks. Experimental validation was conducted using quantitative RT-PCR analysis of key immune markers (TLR2, TLR4, CCR7, IL18) in U937 and K562 leukemia cell lines compared to normal peripheral blood mononuclear cells.</p><p><strong>Results: </strong>The machine learning-derived prognostic model demonstrated exceptional predictive performance with area under the curve values of 0.874, 0.891, and 0.925 for 1-, 2-, and 3-year survival endpoints, respectively. Six critical immune regulatory genes (TLR2, TLR4, CCR7, IL18, TIRAP, FOXP3) were identified as both differentially expressed and prognostically significant, with IL18 showing the highest discriminative capacity (AUC = 0.983). RT-PCR validation confirmed significant upregulation of all tested genes in leukemia cell lines: TLR2 (3.8-fold in U937, 2.2-fold in K562), TLR4 (3.4-fold in U937, 1.8-fold in K562), CCR7 (4.1-fold in U937, 2.7-fold in K562), and IL18 (5.2-fold in U937, 3.6-fold in K562) compared to normal controls (all p < 0.05). Single-cell analysis revealed substantial cellular heterogeneity with cell type-specific expression patterns and complex intercellular communication networks involving B cells, T cells, natural killer cells, and dendritic cells.</p><p><strong>Conclusion: </strong>This study provides a reliable prognostic tool for leukemia and offers insights into the critical role of the immune microenvironment in leukemia pathogenesis. Our findings may guide the development of personalized immunotherapy strategies for leukemia patients.</p>\",\"PeriodicalId\":11148,\"journal\":{\"name\":\"Discover. Oncology\",\"volume\":\"16 1\",\"pages\":\"1846\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-10-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12514105/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Discover. Oncology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s12672-025-03757-9\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Discover. Oncology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s12672-025-03757-9","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Development and validation of a leukemia prognostic model through single-cell RNA sequencing and machine learning approaches.
Background: Leukemia prognosis varies significantly among patients, highlighting the need for accurate prediction tools. Emerging evidence suggests that the immune microenvironment plays a crucial role in leukemia progression and treatment response.
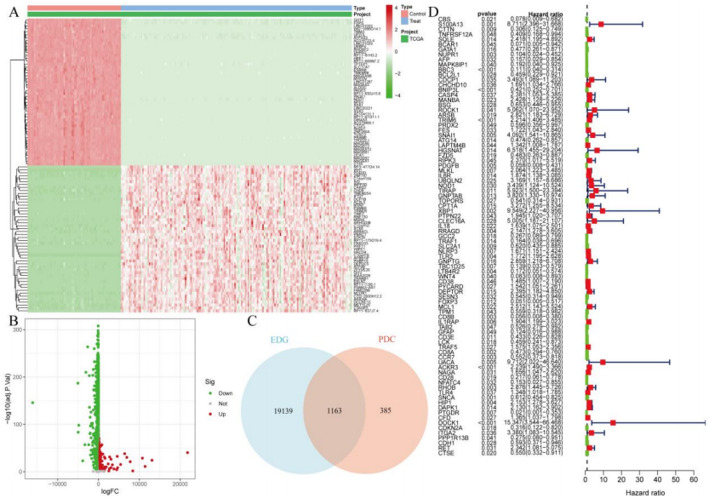
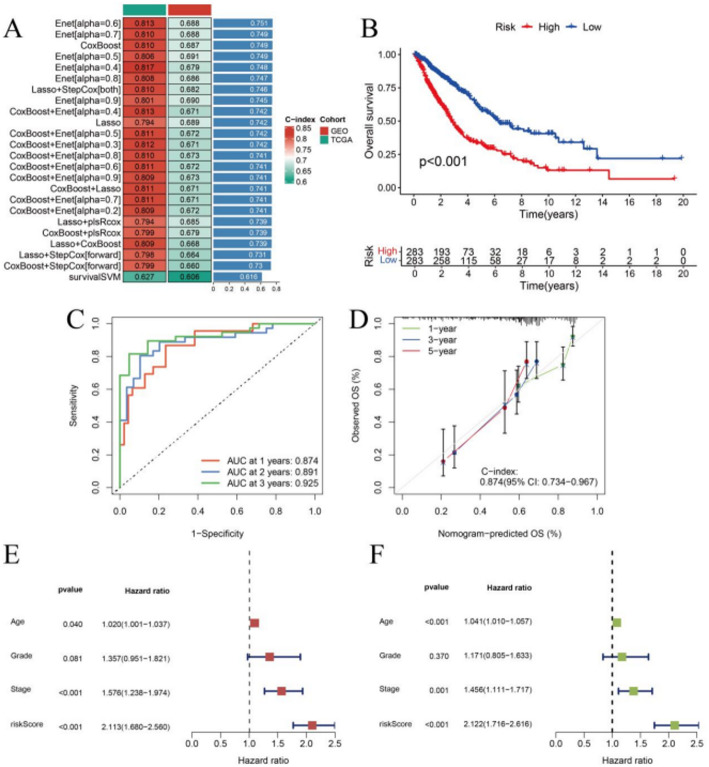
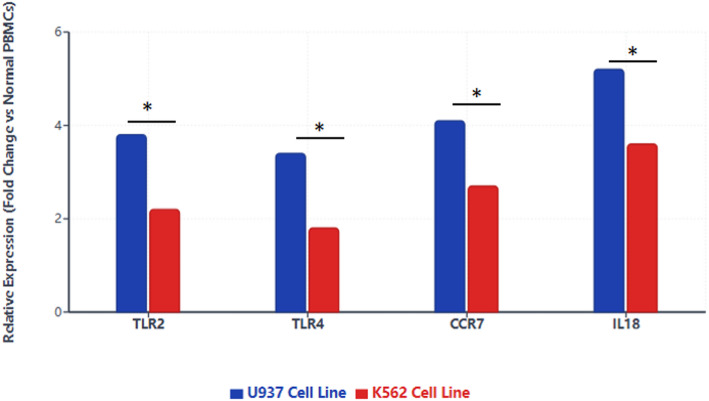
Methods: We analyzed RNA expression profiles and clinical data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases, supplemented by single-cell RNA sequencing datasets. Differential gene expression analysis was performed using stringent criteria (logFC > 1, FDR < 0.05) to identify leukemia-associated genes. Ten distinct machine learning algorithms, including Lasso, CoxBoost, and ensemble methods, were implemented for prognostic model development with cross-platform validation. Single-cell analysis employed Seurat for quality control and cell type annotation, while CellChat algorithm mapped intercellular communication networks. Experimental validation was conducted using quantitative RT-PCR analysis of key immune markers (TLR2, TLR4, CCR7, IL18) in U937 and K562 leukemia cell lines compared to normal peripheral blood mononuclear cells.
Results: The machine learning-derived prognostic model demonstrated exceptional predictive performance with area under the curve values of 0.874, 0.891, and 0.925 for 1-, 2-, and 3-year survival endpoints, respectively. Six critical immune regulatory genes (TLR2, TLR4, CCR7, IL18, TIRAP, FOXP3) were identified as both differentially expressed and prognostically significant, with IL18 showing the highest discriminative capacity (AUC = 0.983). RT-PCR validation confirmed significant upregulation of all tested genes in leukemia cell lines: TLR2 (3.8-fold in U937, 2.2-fold in K562), TLR4 (3.4-fold in U937, 1.8-fold in K562), CCR7 (4.1-fold in U937, 2.7-fold in K562), and IL18 (5.2-fold in U937, 3.6-fold in K562) compared to normal controls (all p < 0.05). Single-cell analysis revealed substantial cellular heterogeneity with cell type-specific expression patterns and complex intercellular communication networks involving B cells, T cells, natural killer cells, and dendritic cells.
Conclusion: This study provides a reliable prognostic tool for leukemia and offers insights into the critical role of the immune microenvironment in leukemia pathogenesis. Our findings may guide the development of personalized immunotherapy strategies for leukemia patients.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


