红外和拉曼光谱,构象稳定性,正坐标分析,内旋势和新型DFT计算脂环1,1-二甲基-1-硅环丁烷
IF 4.7
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
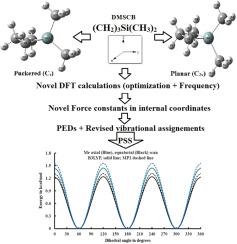
报道了气态和固态的1,1-二甲基-1-硅环丁烷(DMSCB)的中红外光谱(3500 ~ 400 cm-1)。此外,还记录了气相、液相和固相的拉曼光谱(3500 ~ 10 cm-1)。此外,我们还分别对符合C2v/Cs点群的(CH2)3Si(CH3)2分子(DMSCB)进行了量子化学(QC)计算。首先对结构进行优化,然后分别使用6-31G(d)和6-31++G(d,p)基集,采用RHF、B3LYP和MP2方法进行频率计算。初步计算表明皱环构象(Cs)的扭转频率为823-1336 cm−1 (2.352-3.820 kcal/mol),但对于服从C2v点群的构象,得到了一个假想的扭转频率,因此很可能被排除在外。优化后的皱化构象的结构参数与电子衍射技术得到的结构参数具有良好的相关性。利用基于优化结构参数(SPs)的MP2电位表面扫描(PSS),在6-31++G(d,p)基集上的单点能量,采用B3LYP方法估计(CH₃)ₓ/(CH₃)ₑq三重势垒为544/506 cm−1 (1.56/1.45 kcal.mol−1),与460/431 cm−1 (1.32/1.23 kcal.mol−1)相比。利用上述方法估算的红外强度和拉曼活度,将实测的红外和拉曼光谱与模拟的红外和拉曼光谱进行了比较。然而,我们也估计了(CD2)3Si(CH3)2-d6和(CH2)3Si(CD3)2-d6的频率,以证实我们基于亚甲基和甲基基元在氘化作用下的蓝移的解释。因此,我们提出了一种新的基于CS对称的DMSCB谱分析方法,该方法由NCA、内部坐标中的未标度力常数(fc)和势能分布(PEDs)支持。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Infrared and Raman spectra, conformational stability, normal coordinate analysis, barriers to internal rotations and novel DFT calculations of alicyclic 1,1-Dimethyl-1-silacyclobutane
The mid-infrared spectra (3500-400 cm-1) of gaseous and solid 1,1-Dimethyl-1-silacyclobutane (DMSCB) have been reported. Additionally, the Raman spectra (3500-10 cm-1) of the gas, liquid and solid phases have been recorded. Moreover, we have carried out quantum chemical (QC) calculations for (CH2)3Si(CH3)2 molecule (DMSCB) obeying C2v/Cs point group, respectively. Initially the structures are optimized followed by frequency calculations with RHF, B3LYP and MP2 methods using 6-31G(d) and 6-31++G(d,p) basis sets. Preliminary computations favor the puckered ring conformation (Cs) by 823-1336 cm−1 (2.352-3.820 kcal/mol) but an imaginary torsional frequency was obtained for configuration that obeys C2v point group, therefore most likely excluded. The optimized structural parameters (SPs) of the puckered conformer are well correlated to those obtained from electron diffraction techniques. Aided by MP2 potential surface scans (PSS) based on the optimized structural parameters (SPs), single-point energies at 6-31++G(d,p) basis set, the (CH₃)ₐₓ/(CH₃)ₑq three-fold barriers of 544/506 cm−1 (1.56/1.45 kcal.mol−1) were estimated compared to 460/431 cm−1 (1.32/1.23 kcal.mol−1), employing B3LYP method. The observed infrared (IR) and Raman (R) spectra are compared with the simulated IR and Raman spectra using the estimated infrared intensities and the Raman activities, respectively from the above-mentioned methodsy. Nevertheless, frequencies were also estimated for, (CD2)3Si(CH3)2-d6 and (CH2)3Si(CD3)2-d6 to confirm our interpretations based on blue shifts of methylene and methyl fundamentals upon deuteration, respectively. Consequently, we have proposed a new spectral analysis for DMSCB based on CS symmetry, which is supported by NCA, unscaled force constants (FCs) in internal coordinates and potential energy distributions (PEDs).
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊
Journal of Molecular Structure
化学-物理化学
CiteScore
7.10
自引率
15.80%
发文量
2384
审稿时长
45 days
期刊介绍:
The Journal of Molecular Structure is dedicated to the publication of full-length articles and review papers, providing important new structural information on all types of chemical species including:
• Stable and unstable molecules in all types of environments (vapour, molecular beam, liquid, solution, liquid crystal, solid state, matrix-isolated, surface-absorbed etc.)
• Chemical intermediates
• Molecules in excited states
• Biological molecules
• Polymers.
The methods used may include any combination of spectroscopic and non-spectroscopic techniques, for example:
• Infrared spectroscopy (mid, far, near)
• Raman spectroscopy and non-linear Raman methods (CARS, etc.)
• Electronic absorption spectroscopy
• Optical rotatory dispersion and circular dichroism
• Fluorescence and phosphorescence techniques
• Electron spectroscopies (PES, XPS), EXAFS, etc.
• Microwave spectroscopy
• Electron diffraction
• NMR and ESR spectroscopies
• Mössbauer spectroscopy
• X-ray crystallography
• Charge Density Analyses
• Computational Studies (supplementing experimental methods)
We encourage publications combining theoretical and experimental approaches. The structural insights gained by the studies should be correlated with the properties, activity and/ or reactivity of the molecule under investigation and the relevance of this molecule and its implications should be discussed.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


