氟溴席夫碱的晶体学、赫斯菲尔德表面和DFT分析:对结构特征和MAO-A结合电位的见解
IF 4.7
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
本研究合成了一种氟溴取代的席夫碱化合物(E)-2-((2-溴苯基)亚氨基)甲基)-6-氟苯酚(BFSB),并通过实验和理论方法对其结构进行了研究。晶体结构通过单晶x射线衍射确定,揭示了一个正交体系,具有稳定的分子内O-H··N氢键,并通过Hirshfeld表面和能量框架分析证明了卤素和色散相互作用。采用密度泛函理论(DFT)的计算研究提供了与实验几何结构的可靠匹配,并提供了对分子电子结构和反应性的深入了解。此外,尽管存在一些药代动力学限制,ADMET评价预测了良好的药物相似性和易于合成。针对一系列蛋白靶点进行分子对接模拟,探索潜在的生物相互作用,其中MAO-A表现出最强的亲和力。总的来说,结果强调了将晶体学数据与计算建模相结合以表征多功能小分子的相关性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
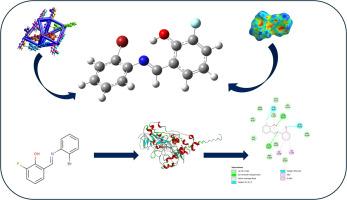
Crystallographic, hirshfeld surface, and DFT analyses of a fluorine–bromine schiff base: Insights into structural features and MAO-A binding potential
In this study, a fluorine–bromine-substituted Schiff base compound, (E)-2-(((2-bromophenyl)imino)methyl)-6-fluorophenol (BFSB), was synthesized and structurally investigated using both experimental and theoretical approaches. The crystal structure, determined by single-crystal X-ray diffraction, revealed an orthorhombic system featuring a stabilizing intramolecular O–H···N hydrogen bond, complemented by halogen and dispersion interactions, as demonstrated through Hirshfeld surface and energy framework analyses. Computational studies employing Density Functional Theory (DFT) provided a reliable match with experimental geometry and offered insight into the molecule’s electronic structure and reactivity. Additionally, ADMET evaluations predicted favorable drug likeness and ease of synthesis despite some pharmacokinetic limitations. Molecular docking simulations were conducted against a range of protein targets to explore potential biological interactions, among which MAO-A showed the strongest affinity. Overall, the results underscore the relevance of combining crystallographic data with computational modeling to characterize multifunctional small molecules.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊
Journal of Molecular Structure
化学-物理化学
CiteScore
7.10
自引率
15.80%
发文量
2384
审稿时长
45 days
期刊介绍:
The Journal of Molecular Structure is dedicated to the publication of full-length articles and review papers, providing important new structural information on all types of chemical species including:
• Stable and unstable molecules in all types of environments (vapour, molecular beam, liquid, solution, liquid crystal, solid state, matrix-isolated, surface-absorbed etc.)
• Chemical intermediates
• Molecules in excited states
• Biological molecules
• Polymers.
The methods used may include any combination of spectroscopic and non-spectroscopic techniques, for example:
• Infrared spectroscopy (mid, far, near)
• Raman spectroscopy and non-linear Raman methods (CARS, etc.)
• Electronic absorption spectroscopy
• Optical rotatory dispersion and circular dichroism
• Fluorescence and phosphorescence techniques
• Electron spectroscopies (PES, XPS), EXAFS, etc.
• Microwave spectroscopy
• Electron diffraction
• NMR and ESR spectroscopies
• Mössbauer spectroscopy
• X-ray crystallography
• Charge Density Analyses
• Computational Studies (supplementing experimental methods)
We encourage publications combining theoretical and experimental approaches. The structural insights gained by the studies should be correlated with the properties, activity and/ or reactivity of the molecule under investigation and the relevance of this molecule and its implications should be discussed.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


