基于度的拓扑指数和机器学习算法的抗高血压药物理化性质预测建模
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
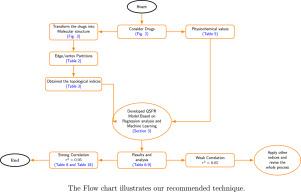
利用分子图理论进行物理化学性质的定量预测已成为化学信息学研究的一个重要热点。本研究引入一套基于度的拓扑指标abc、ABS、MMR、SDD、SI、SO、SO3、so4对23种降压药物进行建模。使用经典线性回归和基于集成的机器学习算法(Random Forest和XGBoost)开发了QSPR框架。使用标准误差指标(MAE, MSE, RMSE, R2)评估模型性能,并通过基尼系数,排列和Shapley加性解释(SHAP)分析特征重要性。所提出的指标与沸点、熔点、临界体积、LogP、摩尔折射率和CLogP有很强的相关性。在测试的模型中,XGBoost表现最好,在所有属性中实现R2>;0.99。除了预测准确性之外,研究结果表明,基于度的指数捕捉了药物分子的结构特征,同时为亲脂性、稳定性和热力学行为提供了可解释的见解。这些结果证明了图形理论描述符作为具有成本效益的实验分析替代方案的潜力,从而加速了合理的药物设计和筛选工作流程。总的来说,本研究建立了一个可推广的模型框架,连接数学化学和药物应用,为高通量药物发现提供了有价值的方向。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Predictive modeling of physicochemical properties of antihypertensive drugs using degree-based topological indices and machine learning algorithm
Quantitative prediction of physicochemical properties through molecular graph theory has become an important focus in cheminformatics. This study introduces a set of degree-based topological indices—ABC, ABS, MMR, SDD, SI, SO, SO, and SO—to model 23 antihypertensive drugs. A QSPR framework is developed using both classical linear regression and ensemble-based machine learning algorithms (Random Forest and XGBoost). Model performance is evaluated using standard error metrics (MAE, MSE, RMSE, ), and feature importance is analyzed through Gini, permutation, and Shapley Additive exPlanations (SHAP). The proposed indices show strong correlations with boiling point, melting point, critical volume, LogP, molar refractivity, and CLogP. Among the tested models, XGBoost performs best, achieving across all properties. Beyond predictive accuracy, the findings show that degree-based indices capture structural features of drug molecules while offering interpretable insights into lipophilicity, stability, and thermodynamic behavior. These results demonstrate the potential of graph-theoretical descriptors as cost-effective alternatives to experimental assays, thereby accelerating rational drug design and screening workflows. Overall, this study establishes a generalizable modeling framework that bridges mathematical chemistry and pharmaceutical applications, providing valuable directions for high-throughput drug discovery.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


