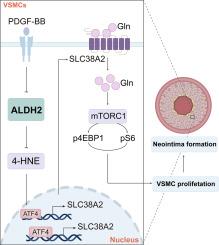
ALDH2缺乏通过slc38a2介导的谷氨酰胺摄取上调,增强血管平滑肌细胞增殖,从而加重血管损伤诱导的再狭窄。
IF 11.9
1区 医学
Q1 ENDOCRINOLOGY & METABOLISM
引用次数: 0
摘要
背景与目的:血管损伤性再狭窄是冠状动脉疾病(CAD)患者长期预后不良的重要原因。尽管醛脱氢酶2 (ALDH2)缺乏与CAD患者预后不良有关,但ALDH2影响血管损伤诱导的再狭窄的确切机制仍不清楚。在此,我们试图探讨ALDH2在调节血管平滑肌细胞(VSMC)增殖和血管损伤诱导的再狭窄中的作用。方法和结果:免疫荧光和免疫印迹显示,人冠状动脉狭窄段和损伤小鼠股动脉、颈动脉VSMCs中ALDH2表达明显降低。整体敲除ALDH2和vsmc特异性敲除ALDH2加剧了损伤诱导的新内膜形成,而vsmc特异性ALDH2过表达则减少了新内膜的形成。内皮细胞(EC)特异性ALDH2敲除对损伤诱导的新内膜形成影响不大。机制研究表明,ALDH2缺乏通过上调谷氨酰胺转运蛋白SLC38A2的表达促进VSMC增殖,SLC38A2是一个新的ALDH2靶基因。进一步的生物信息学分析、荧光素酶测定和ChIP-qPCR显示,ALDH2缺乏通过激活转录因子4 (ATF4)增加SLC38A2的表达,ATF4敲低在很大程度上逆转了ALDH2缺乏促进VSMC增殖的能力。此外,ALDH2缺乏促进了4-HNE内合蛋白的积累,从而激活ATF4,从而增加了vsmc中SLC28A2的转录活性。重要的是,通过腺相关病毒血清2型(AAV2) shRNA或抑制剂MeAIB下调SLC38A2在限制VSMC增殖和新内膜形成方面具有良好的治疗潜力。最后,我们证明了ALDH2E506k突变小鼠VSMC增殖加剧,新生内膜形成。结论:我们的研究阐明了ALDH2缺乏通过增加谷氨酰胺摄取来促进VSMC增殖,从而加剧新内膜形成的新机制,为预防血管损伤性再狭窄提供了一个有希望的翻译策略。本文章由计算机程序翻译,如有差异,请以英文原文为准。
ALDH2 deficiency aggravates vascular injury-induced restenosis by enhancing vascular smooth muscle cell proliferation through SLC38A2-mediated upregulation of glutamine uptake
Background and aims
Vascular injury-induced restenosis is an important cause of poor long-term prognosis in patients with coronary artery disease (CAD). Although aldehyde dehydrogenase 2 (ALDH2) deficiency has been linked to poor outcomes in CAD patients, the precise mechanisms through which ALDH2 influences vascular injury-induced restenosis remain elusive. Herein, we attempted to explore the role of ALDH2 in modulating vascular smooth muscle cell (VSMC) proliferation and vascular injury-induced restenosis.
Methods and results
Immunofluorescence and immunoblotting revealed that ALDH2 expression was significantly decreased in VSMCs in human stenotic coronary segments and injured mouse femoral and carotid arteries. Global ALDH2 knockout and VSMC-specific ALDH2 knockout exacerbated injury-induced neointima formation, whereas VSMC-specific ALDH2 overexpression reduced neointima formation. Endothelial cell (EC)-specific ALDH2 knockout had little effect on injury-induced neointima formation. Mechanistic studies revealed that ALDH2 deficiency facilitated VSMC proliferation by upregulating the expression of the glutamine transporter SLC38A2, which is a novel ALDH2 target gene. Further bioinformatics analysis, luciferase assays, and ChIP–qPCR revealed that ALDH2 deficiency increased SLC38A2 expression via activating transcription factor 4 (ATF4) and that ATF4 knockdown largely reversed the ability of ALDH2 deficiency to promote VSMC proliferation. Moreover, ALDH2 deficiency promoted the accumulation of 4-HNE adducted proteins, thereby activating ATF4, which subsequently increased SLC28A2 transcriptional activity in VSMCs. Importantly, downregulation of SLC38A2 by adeno-associated virus serotype 2 (AAV2) shRNA or by the inhibitor MeAIB has promising therapeutic potential in limiting VSMC proliferation and neointima formation. Finally, we demonstrated that VSMC proliferation was aggravated and that neointima formation occurred in ALDH2E506k mutant mice.
Conclusion
Our study elucidates a novel mechanism through which ALDH2 deficiency aggravates neointimal formation by enhancing VSMC proliferation through an increase in glutamine uptake, suggesting a promising translational strategy for the prevention of vascular injury-induced restenosis.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Metabolism: clinical and experimental
医学-内分泌学与代谢
CiteScore
18.90
自引率
3.10%
发文量
310
审稿时长
16 days
期刊介绍:
Metabolism upholds research excellence by disseminating high-quality original research, reviews, editorials, and commentaries covering all facets of human metabolism.
Consideration for publication in Metabolism extends to studies in humans, animal, and cellular models, with a particular emphasis on work demonstrating strong translational potential.
The journal addresses a range of topics, including:
- Energy Expenditure and Obesity
- Metabolic Syndrome, Prediabetes, and Diabetes
- Nutrition, Exercise, and the Environment
- Genetics and Genomics, Proteomics, and Metabolomics
- Carbohydrate, Lipid, and Protein Metabolism
- Endocrinology and Hypertension
- Mineral and Bone Metabolism
- Cardiovascular Diseases and Malignancies
- Inflammation in metabolism and immunometabolism
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


