{"title":"OTMol:基于最优输运的鲁棒分子结构比较。","authors":"Xiaoqi Wei, Xuhang Dai, Yaqi Wu, Yanxiang Zhao, Yingkai Zhang, Zixuan Cang","doi":"10.1021/acs.jcim.5c02099","DOIUrl":null,"url":null,"abstract":"<p><p>Root-mean-square deviation (RMSD) is widely used to assess structural similarity in systems ranging from flexible ligand conformers to complex molecular cluster configurations. Despite its wide utility, the RMSD calculation is often challenged by inconsistent atom ordering, indistinguishable configurations in molecular clusters, and potential chirality inversion during alignment. These issues highlight the necessity of accurately establishing atom-to-atom correspondence as a prerequisite for meaningful alignment. Traditional approaches often rely on heuristic cost matrices combined with the Hungarian algorithm, yet these methods underutilize the rich intramolecular structural information and may fail to generalize across chemically diverse systems. In this work, we introduce OTMol, a method that formulates the molecular alignment task as a fused supervised Gromov-Wasserstein (fsGW) optimal transport problem. By leveraging the intrinsic geometric and topological relationships within each molecule, we find that OTMol eliminates the need for manually defined cost functions and enables a principled, data-driven matching strategy. Importantly, OTMol preserves key chemical features, such as molecular chirality and bond connectivity consistency. We evaluate OTMol across a wide range of molecular systems, including adenosine triphosphate, imatinib, lipids, small peptides, and water clusters, and demonstrate that it consistently achieves low RMSD values while preserving computational efficiency. Importantly, the synthesis of OTMol maintains molecular integrity by enforcing one-to-one mappings between entire molecules, thereby avoiding erroneous many-to-one alignments that often arise in comparing molecular clusters. Our results underscore the utility of optimal transport theory for molecular alignment and offer a generalizable framework applicable to structural comparison tasks in cheminformatics, molecular modeling, and related disciplines.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2025-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"OTMol: Robust Molecular Structure Comparison via Optimal Transport.\",\"authors\":\"Xiaoqi Wei, Xuhang Dai, Yaqi Wu, Yanxiang Zhao, Yingkai Zhang, Zixuan Cang\",\"doi\":\"10.1021/acs.jcim.5c02099\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Root-mean-square deviation (RMSD) is widely used to assess structural similarity in systems ranging from flexible ligand conformers to complex molecular cluster configurations. Despite its wide utility, the RMSD calculation is often challenged by inconsistent atom ordering, indistinguishable configurations in molecular clusters, and potential chirality inversion during alignment. These issues highlight the necessity of accurately establishing atom-to-atom correspondence as a prerequisite for meaningful alignment. Traditional approaches often rely on heuristic cost matrices combined with the Hungarian algorithm, yet these methods underutilize the rich intramolecular structural information and may fail to generalize across chemically diverse systems. In this work, we introduce OTMol, a method that formulates the molecular alignment task as a fused supervised Gromov-Wasserstein (fsGW) optimal transport problem. By leveraging the intrinsic geometric and topological relationships within each molecule, we find that OTMol eliminates the need for manually defined cost functions and enables a principled, data-driven matching strategy. Importantly, OTMol preserves key chemical features, such as molecular chirality and bond connectivity consistency. We evaluate OTMol across a wide range of molecular systems, including adenosine triphosphate, imatinib, lipids, small peptides, and water clusters, and demonstrate that it consistently achieves low RMSD values while preserving computational efficiency. Importantly, the synthesis of OTMol maintains molecular integrity by enforcing one-to-one mappings between entire molecules, thereby avoiding erroneous many-to-one alignments that often arise in comparing molecular clusters. Our results underscore the utility of optimal transport theory for molecular alignment and offer a generalizable framework applicable to structural comparison tasks in cheminformatics, molecular modeling, and related disciplines.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-10-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.5c02099\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.5c02099","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
OTMol: Robust Molecular Structure Comparison via Optimal Transport.
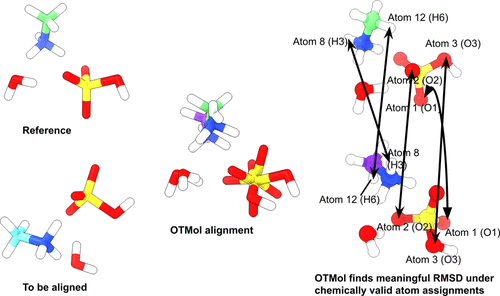
Root-mean-square deviation (RMSD) is widely used to assess structural similarity in systems ranging from flexible ligand conformers to complex molecular cluster configurations. Despite its wide utility, the RMSD calculation is often challenged by inconsistent atom ordering, indistinguishable configurations in molecular clusters, and potential chirality inversion during alignment. These issues highlight the necessity of accurately establishing atom-to-atom correspondence as a prerequisite for meaningful alignment. Traditional approaches often rely on heuristic cost matrices combined with the Hungarian algorithm, yet these methods underutilize the rich intramolecular structural information and may fail to generalize across chemically diverse systems. In this work, we introduce OTMol, a method that formulates the molecular alignment task as a fused supervised Gromov-Wasserstein (fsGW) optimal transport problem. By leveraging the intrinsic geometric and topological relationships within each molecule, we find that OTMol eliminates the need for manually defined cost functions and enables a principled, data-driven matching strategy. Importantly, OTMol preserves key chemical features, such as molecular chirality and bond connectivity consistency. We evaluate OTMol across a wide range of molecular systems, including adenosine triphosphate, imatinib, lipids, small peptides, and water clusters, and demonstrate that it consistently achieves low RMSD values while preserving computational efficiency. Importantly, the synthesis of OTMol maintains molecular integrity by enforcing one-to-one mappings between entire molecules, thereby avoiding erroneous many-to-one alignments that often arise in comparing molecular clusters. Our results underscore the utility of optimal transport theory for molecular alignment and offer a generalizable framework applicable to structural comparison tasks in cheminformatics, molecular modeling, and related disciplines.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


