CO2电还原Ni-N-C催化剂中真正活性位点的揭示
IF 15.6
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
理解和设计单原子催化剂(SACs)的活性位点需要超越静态模型来捕捉它们在实际电化学条件下的动态演变。在这里,我们通过将大正则密度泛函理论(GC-DFT)与机器学习加速采样相结合,开发了一个考虑操作条件的综合理论框架,以揭示Ni-N-C SACs中用于CO2还原反应(CO2RR)的结构-活性-稳定性关系。系统地评价了NiNxC4-x (x = 0-4)基序库,这些基序代表了高温合成过程中可能形成的配位缺陷。在工作条件下,发现这些位点发生了氢化反应,NiN3C1_H1被确定为最可能的活性位点。在还原电位下,氢自发地吸附在C-Ni桥位点而不是Ni顶部位点,而地下氢促进了对活化至关重要的弯曲CO2吸附。CO2RR对CO的高选择性源于位点分离:Ni中心驱动CO2RR,而析氢反应(HER)发生在C-Ni桥或N位点,以及适度氢覆盖下HER的热力学抑制。在更多的负电位下,CO2RR速率决定过程(RDP)的变化和H和H2O的共吸附引起的Ni的表面外位移共同降低了活性和选择性。因此,Ni-N-C催化剂的高CO2RR选择性及其负电位反转都可以通过考虑氢化表面来解释。这凸显了造型逼真的必要性;在原位条件下。该框架提供了对SACs活性位点动态行为的一般性见解,为广泛电化学反应的活性和稳健催化剂的合理设计提供了指导。本文章由计算机程序翻译,如有差异,请以英文原文为准。
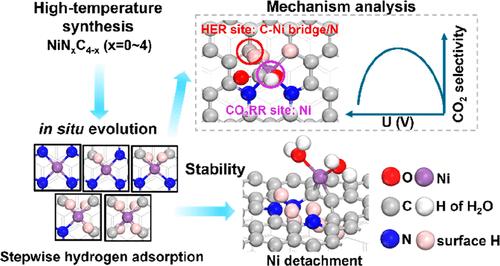
Uncovering the True Active Sites in Ni–N–C Catalysts for CO2 Electroreduction
Understanding and designing active sites in single-atom catalysts (SACs) requires going beyond static models to capture their dynamic evolution under realistic electrochemical conditions. Here, we develop an integrated theoretical framework that accounts for operational conditions, by combining grand canonical density functional theory (GC-DFT) with machine-learning-accelerated sampling, to uncover structure–activity–stability relationships in Ni–N–C SACs for the CO2 reduction reaction (CO2RR). A library of NiNxC4–x (x = 0–4) motifs─representing coordination defects likely formed during high-temperature synthesis─was systematically evaluated. Under working conditions, these sites were found to undergo hydrogenation, and NiN3C1_H1 was identified as the most probable active site. At reducing potentials, hydrogen adsorbs spontaneously at C–Ni bridge sites rather than Ni top sites, while subsurface hydrogen facilitates bent CO2 adsorption crucial for activation. High CO2RR selectivity toward CO arises from site separation: Ni centers drive CO2RR, while the hydrogen evolution reaction (HER) occurs at the C–Ni bridge or N sites and from thermodynamic suppression of HER at moderate hydrogen coverage. At more negative potentials, a shift in the CO2RR rate-determining process (RDP) and Ni out-of-surface displacement induced by coadsorption of H and H2O jointly reduce activity and selectivity. Thus, both the high CO2RR selectivity of Ni–N–C catalysts and its reversal with more negative potentials can be rationalized by accounting for hydrogenated surfaces. This highlights the necessity of modeling realistic; in situ conditions. This framework provides generalizable insights into the dynamic behavior of active sites in SACs, offering guidance for the rational design of active and robust catalysts for a wide range of electrochemical reactions.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


