通过从头算分子动力学和z熵理论实现精确的熵和熔点:在氟和氯化物熔盐中的应用
IF 5.2
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
我们最近开发了一种突破性的方法,通过将多尺度熵方法(称为z熵理论)与分子动力学(MD)模拟相结合,快速计算固体和液体中的熵。这种方法通过分析局部结构构型和原子分布的概率,可以从单个MD轨迹中进行熵估计,有效地解决了长期以来捕获构型熵的挑战,特别是对于液体。在这里,我们通过使用从头算MD (AIMD)模拟来预测25种二元和三元氟化物和氯化物熔盐的熵、焓和熔点,从而证明了该方法的功能。我们的预测和实验数据之间的显著一致性强调了这种方法在改变计算热力学方面的潜力,为固体和液体的热力学性质提供准确、有效和直接的预测。本文章由计算机程序翻译,如有差异,请以英文原文为准。
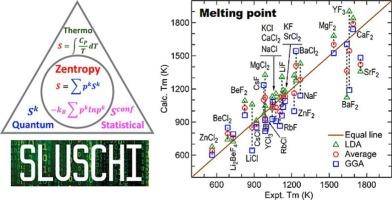
Achieving accurate entropy and melting point by ab initio molecular dynamics and zentropy theory: Application to fluoride and chloride molten salts
We have recently developed a breakthrough methodology for rapidly computing entropy in both solids and liquids by integrating a multiscale entropy approach (known as zentropy theory) with molecular dynamics (MD) simulations. This approach enables entropy estimation from a single MD trajectory by analyzing the probabilities of local structural configurations and atomic distributions, effectively addressing the long-standing challenge of capturing configurational entropy, particularly for liquid. Here, we demonstrate the power of this method by predicting entropies, enthalpies, and melting points of 25 binary and ternary fluoride- and chloride-based molten salts using ab initio MD (AIMD) simulations. The remarkable agreement between our predictions and experimental data underscores the potential of this approach to transform computational thermodynamics, offering accurate, efficient, and direct predictions of thermodynamic properties across both solid and liquid phases.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Molecular Liquids
化学-物理:原子、分子和化学物理
CiteScore
10.30
自引率
16.70%
发文量
2597
审稿时长
78 days
期刊介绍:
The journal includes papers in the following areas:
– Simple organic liquids and mixtures
– Ionic liquids
– Surfactant solutions (including micelles and vesicles) and liquid interfaces
– Colloidal solutions and nanoparticles
– Thermotropic and lyotropic liquid crystals
– Ferrofluids
– Water, aqueous solutions and other hydrogen-bonded liquids
– Lubricants, polymer solutions and melts
– Molten metals and salts
– Phase transitions and critical phenomena in liquids and confined fluids
– Self assembly in complex liquids.– Biomolecules in solution
The emphasis is on the molecular (or microscopic) understanding of particular liquids or liquid systems, especially concerning structure, dynamics and intermolecular forces. The experimental techniques used may include:
– Conventional spectroscopy (mid-IR and far-IR, Raman, NMR, etc.)
– Non-linear optics and time resolved spectroscopy (psec, fsec, asec, ISRS, etc.)
– Light scattering (Rayleigh, Brillouin, PCS, etc.)
– Dielectric relaxation
– X-ray and neutron scattering and diffraction.
Experimental studies, computer simulations (MD or MC) and analytical theory will be considered for publication; papers just reporting experimental results that do not contribute to the understanding of the fundamentals of molecular and ionic liquids will not be accepted. Only papers of a non-routine nature and advancing the field will be considered for publication.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


