Arindam Ghosh, Saba Annigeri, Chakita Singh, Sunil Kumar Hemram
{"title":"双‘A’型伴矿化皮质激素缺乏:Allgrove综合征的罕见表现。","authors":"Arindam Ghosh, Saba Annigeri, Chakita Singh, Sunil Kumar Hemram","doi":"10.1297/cpe.2025-0015","DOIUrl":null,"url":null,"abstract":"<p><p>Allgrove syndrome (AS), an uncommon multisystem disorder, is characterized by the classic clinical triad of alacrimia, achalasia, and adrenal insufficiency, and is typically limited to glucocorticoid deficiency with preserved mineralocorticoid (MC) function. Here, we present the case of a 5-yr-old girl with alacrimia since birth, failure to thrive, and generalized hyperpigmentation for the past two years, who presented to the emergency department with an altered sensorium. Upon admission, the patient was found to have hypoglycemia and hyponatremia. After subsequent evaluation, the patient was diagnosed with phenotypically incomplete AS with mineralocorticoid insufficiency and harbored a novel homozygous mutation in exon 7 of the <i>AAAS</i> gene (c.618del; p.Ser207LeufsTer84). Treatment with hydrocortisone and fludrocortisone yielded remarkable outcomes. Given the variable presentations of this condition, a high index of clinical suspicion and awareness of atypical features are essential for early diagnosis and initiation of coordinated care to prevent unnecessary morbidity and mortality. When AS is suspected, molecular genetic testing should be performed to confirm the diagnosis, plan management, and provide genetic counseling.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":"34 4","pages":"254-259"},"PeriodicalIF":1.2000,"publicationDate":"2025-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12494399/pdf/","citationCount":"0","resultStr":"{\"title\":\"Double 'A' phenotypes with mineralocorticoid deficiency: A rare presentation of Allgrove syndrome.\",\"authors\":\"Arindam Ghosh, Saba Annigeri, Chakita Singh, Sunil Kumar Hemram\",\"doi\":\"10.1297/cpe.2025-0015\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Allgrove syndrome (AS), an uncommon multisystem disorder, is characterized by the classic clinical triad of alacrimia, achalasia, and adrenal insufficiency, and is typically limited to glucocorticoid deficiency with preserved mineralocorticoid (MC) function. Here, we present the case of a 5-yr-old girl with alacrimia since birth, failure to thrive, and generalized hyperpigmentation for the past two years, who presented to the emergency department with an altered sensorium. Upon admission, the patient was found to have hypoglycemia and hyponatremia. After subsequent evaluation, the patient was diagnosed with phenotypically incomplete AS with mineralocorticoid insufficiency and harbored a novel homozygous mutation in exon 7 of the <i>AAAS</i> gene (c.618del; p.Ser207LeufsTer84). Treatment with hydrocortisone and fludrocortisone yielded remarkable outcomes. Given the variable presentations of this condition, a high index of clinical suspicion and awareness of atypical features are essential for early diagnosis and initiation of coordinated care to prevent unnecessary morbidity and mortality. When AS is suspected, molecular genetic testing should be performed to confirm the diagnosis, plan management, and provide genetic counseling.</p>\",\"PeriodicalId\":10678,\"journal\":{\"name\":\"Clinical Pediatric Endocrinology\",\"volume\":\"34 4\",\"pages\":\"254-259\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2025-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12494399/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pediatric Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1297/cpe.2025-0015\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/7/17 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2025-0015","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/7/17 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Double 'A' phenotypes with mineralocorticoid deficiency: A rare presentation of Allgrove syndrome.
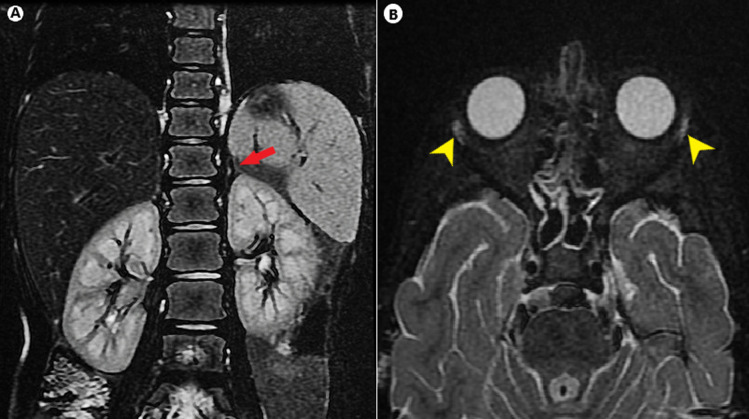
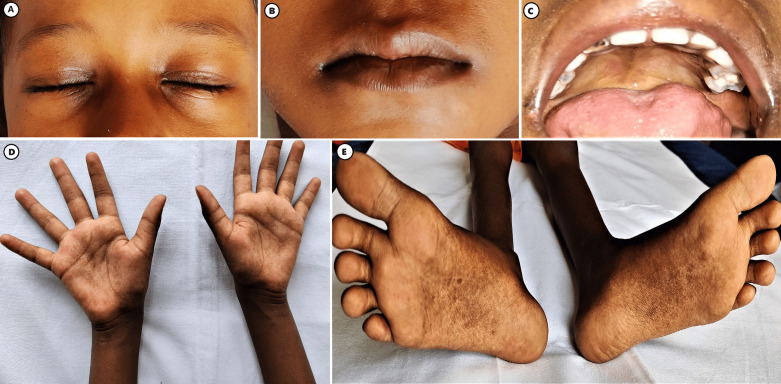
Allgrove syndrome (AS), an uncommon multisystem disorder, is characterized by the classic clinical triad of alacrimia, achalasia, and adrenal insufficiency, and is typically limited to glucocorticoid deficiency with preserved mineralocorticoid (MC) function. Here, we present the case of a 5-yr-old girl with alacrimia since birth, failure to thrive, and generalized hyperpigmentation for the past two years, who presented to the emergency department with an altered sensorium. Upon admission, the patient was found to have hypoglycemia and hyponatremia. After subsequent evaluation, the patient was diagnosed with phenotypically incomplete AS with mineralocorticoid insufficiency and harbored a novel homozygous mutation in exon 7 of the AAAS gene (c.618del; p.Ser207LeufsTer84). Treatment with hydrocortisone and fludrocortisone yielded remarkable outcomes. Given the variable presentations of this condition, a high index of clinical suspicion and awareness of atypical features are essential for early diagnosis and initiation of coordinated care to prevent unnecessary morbidity and mortality. When AS is suspected, molecular genetic testing should be performed to confirm the diagnosis, plan management, and provide genetic counseling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


