从从头算分子动力学确定最大熵势
IF 2.9
4区 工程技术
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
摘要
在原子水平上理解电化学界面对于优化能量转换和存储技术的催化性能至关重要。本文介绍了一种基于从头算分子动力学(AIMD)模拟的计算框架,用于预测双电层无序和电荷转移效率的最大熵势(PME)。通过将AIMD与广义计算氢电极相结合,系统地研究了电解质组成、阳离子同一性和pH对PME位置的影响。该方法重现了Au和Pt电极PME的实验变化,并为混合阳离子体系中多个PME值的出现提供了前所未有的见解。研究结果通过揭示混合阳离子体系中多个PME值的存在,挑战了传统的电解质结构模型。这表明阳离子、吸附剂和界面紊乱之间的相互作用比以前假设的更为复杂。本研究开发的计算框架为理解这些相互作用提供了预测工具,为调整电催化活性提供了新的策略。本文章由计算机程序翻译,如有差异,请以英文原文为准。
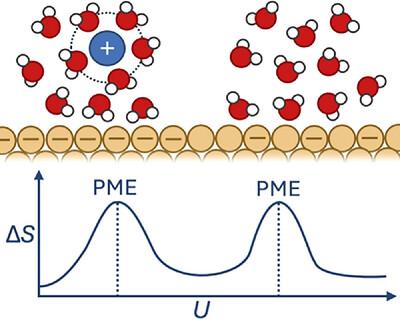
Determining the Potential of Maximum Entropy from Ab Initio Molecular Dynamics
Understanding electrochemical interfaces at the atomic level is essential for optimizing catalytic performance in energy conversion and storage technologies. This study introduces a computational framework based on ab initio molecular dynamics (AIMD) simulations to predict the potential of maximum entropy (PME) a descriptor of electric double layer disorder and charge transfer efficiency. By integrating AIMD with the generalized computational hydrogen electrode, it is systematically investigated how electrolyte composition, cation identity, and pH effect the position of PME. The approach reproduces experimental shifts in PME for Au and Pt electrodes and provides unprecedented insights into the emergence of multiple PME values in mixed-cation systems. The findings challenge conventional models of electrolyte structuring by revealing the presence of multiple PME values within mixed-cation systems. This suggests a more complex interplay between cations, adsorbates, and interfacial disorder than previously assumed. The computational framework developed in this study provides a predictive tool for understanding these interactions, offering new strategies for tuning electrocatalytic activity.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Advanced Theory and Simulations
Multidisciplinary-Multidisciplinary
CiteScore
5.50
自引率
3.00%
发文量
221
期刊介绍:
Advanced Theory and Simulations is an interdisciplinary, international, English-language journal that publishes high-quality scientific results focusing on the development and application of theoretical methods, modeling and simulation approaches in all natural science and medicine areas, including:
materials, chemistry, condensed matter physics
engineering, energy
life science, biology, medicine
atmospheric/environmental science, climate science
planetary science, astronomy, cosmology
method development, numerical methods, statistics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


