{"title":"有机分子晶体中电荷-能量输运态密度比例的熵律法计算研究","authors":"Muthuvel Pavalamuthu, Karuppuchamy Navamani","doi":"10.1021/acs.jpcc.5c04381","DOIUrl":null,"url":null,"abstract":"The charge-energy transport mechanism in molecules is of fundamental importance and practical interest in the design of highly efficient molecular electronic devices. Importantly, the production of entropy due to charge dynamics-associated energy flux variation in the molecular solids causes nonequilibrium transport, and it leads to physics of deviation in Einstein’s diffusion-mobility relation of <i></i><math display=\"inline\"><mfrac><mi>D</mi><mi>μ</mi></mfrac><mo>=</mo><mfrac><mrow><msub><mi>k</mi><mi mathvariant=\"normal\">B</mi></msub><mi>T</mi></mrow><mi>q</mi></mfrac></math> (Hurowitz, D.; Cohen, D., <i>Phys. Rev. E</i> <b>2014</b>, 90, No. 032129 and Navamani, K., <i>J. Phys. Chem. Lett.</i> <b>2025</b>, 16, 8596–8612). This urges the requirement of a suitable alternative method to attain better accuracy in semiconducting and optoelectronic properties in different molecular devices. With this motivation, the “density of states (DOS) proportion” is proposed to incorporate a band and nonadiabatic hopping transport mechanism at different thermodynamic conditions. The DOS proportion is in direct proportion with the population of electronic states or normal DOS. In this context, the developed <i>D</i>/μ relation and other extended charge transport (CT) formalism (conductivity, current density, etc.) are used to study the CT in dialkyl substituted dithienothiophene (<b>DSDTT</b>)-based molecular crystals. The CT key parameters such as charge transfer integral, site energy, and spatial overlap integral are computed from the dimer projection (DIPRO) method using electronic structure calculations. These CT key parameters, including reorganization energy, are used in the degeneracy-weighted diffusion equation and the DOS proportion factor to explore the CT in the <b>DSDTT</b>-based molecular derivatives. Using the entropy-ruled method under degenerate and large carrier energy flux approximation, the calculated hole mobility values are around 0.85 and 0.19 cm<sup>2</sup>/V s for <b>DSDTT-1</b> and <b>DSDTT-2</b> molecular crystals, respectively, reaching a proximity value with an experimentally obtained average mobility. From the obtained ideality factor from the quantum-corrected Shockley diode equation, Langevin transport (trap-free diffusion) is observed in these <b>DSDTT</b> molecules, which suggests that these molecules are good candidates for designing hole transporting (or P-type semiconducting) devices.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"46 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2025-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Entropy-Ruled Method on Density of States Proportion for Charge-Energy Transport in Organic Molecular Crystals: A Computational Study\",\"authors\":\"Muthuvel Pavalamuthu, Karuppuchamy Navamani\",\"doi\":\"10.1021/acs.jpcc.5c04381\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The charge-energy transport mechanism in molecules is of fundamental importance and practical interest in the design of highly efficient molecular electronic devices. Importantly, the production of entropy due to charge dynamics-associated energy flux variation in the molecular solids causes nonequilibrium transport, and it leads to physics of deviation in Einstein’s diffusion-mobility relation of <i></i><math display=\\\"inline\\\"><mfrac><mi>D</mi><mi>μ</mi></mfrac><mo>=</mo><mfrac><mrow><msub><mi>k</mi><mi mathvariant=\\\"normal\\\">B</mi></msub><mi>T</mi></mrow><mi>q</mi></mfrac></math> (Hurowitz, D.; Cohen, D., <i>Phys. Rev. E</i> <b>2014</b>, 90, No. 032129 and Navamani, K., <i>J. Phys. Chem. Lett.</i> <b>2025</b>, 16, 8596–8612). This urges the requirement of a suitable alternative method to attain better accuracy in semiconducting and optoelectronic properties in different molecular devices. With this motivation, the “density of states (DOS) proportion” is proposed to incorporate a band and nonadiabatic hopping transport mechanism at different thermodynamic conditions. The DOS proportion is in direct proportion with the population of electronic states or normal DOS. In this context, the developed <i>D</i>/μ relation and other extended charge transport (CT) formalism (conductivity, current density, etc.) are used to study the CT in dialkyl substituted dithienothiophene (<b>DSDTT</b>)-based molecular crystals. The CT key parameters such as charge transfer integral, site energy, and spatial overlap integral are computed from the dimer projection (DIPRO) method using electronic structure calculations. These CT key parameters, including reorganization energy, are used in the degeneracy-weighted diffusion equation and the DOS proportion factor to explore the CT in the <b>DSDTT</b>-based molecular derivatives. Using the entropy-ruled method under degenerate and large carrier energy flux approximation, the calculated hole mobility values are around 0.85 and 0.19 cm<sup>2</sup>/V s for <b>DSDTT-1</b> and <b>DSDTT-2</b> molecular crystals, respectively, reaching a proximity value with an experimentally obtained average mobility. From the obtained ideality factor from the quantum-corrected Shockley diode equation, Langevin transport (trap-free diffusion) is observed in these <b>DSDTT</b> molecules, which suggests that these molecules are good candidates for designing hole transporting (or P-type semiconducting) devices.\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"46 1\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-10-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpcc.5c04381\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.5c04381","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
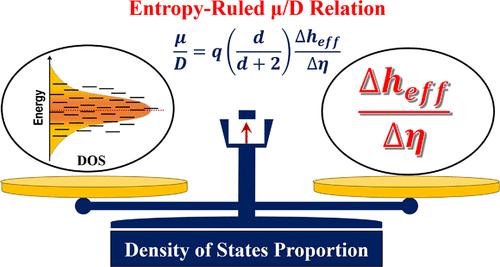
分子中的电荷-能量输运机制在高效分子电子器件的设计中具有重要的基础和实际意义。重要的是,由于分子固体中电荷动力学相关的能量通量变化而产生的熵导致非平衡输运,并导致爱因斯坦的扩散-迁移率关系Dμ=kBTq的物理偏差(Hurowitz, D.; Cohen, D., Phys.)。中国生物医学工程学报,2014,32(2):532 - 529。化学。生态学报,2015,16,8596-8612)。这促使人们需要一种合适的替代方法,以在不同的分子器件中获得更好的半导体和光电子特性精度。基于这一动机,提出了“态密度(DOS)比例”,该比例包含了不同热力学条件下的带和非绝热跳输运机制。DOS比例与电子态总体或正常DOS成正比。在此背景下,利用D/μ关系式和其他扩展电荷输运(CT)形式(电导率、电流密度等)研究了二烷基取代二噻吩(DSDTT)基分子晶体中的电荷输运(CT)。利用二聚体投影(DIPRO)方法,利用电子结构计算得到电荷转移积分、位能和空间重叠积分等CT关键参数。将重组能等CT关键参数应用于简并加权扩散方程和DOS比例因子中,探讨dsdtt基分子衍生物中的CT。在简并和大载流子能量通量近似下,利用熵律法计算得到的DSDTT-1和DSDTT-2分子晶体的空穴迁移率分别在0.85和0.19 cm2/V s左右,与实验得到的平均迁移率接近。从量子校正的肖克利二极管方程得到的理想因子中,在这些DSDTT分子中观察到朗格万输运(无阱扩散),这表明这些分子是设计空穴输运(或p型半导体)器件的良好候选者。
Entropy-Ruled Method on Density of States Proportion for Charge-Energy Transport in Organic Molecular Crystals: A Computational Study
The charge-energy transport mechanism in molecules is of fundamental importance and practical interest in the design of highly efficient molecular electronic devices. Importantly, the production of entropy due to charge dynamics-associated energy flux variation in the molecular solids causes nonequilibrium transport, and it leads to physics of deviation in Einstein’s diffusion-mobility relation of (Hurowitz, D.; Cohen, D., Phys. Rev. E2014, 90, No. 032129 and Navamani, K., J. Phys. Chem. Lett.2025, 16, 8596–8612). This urges the requirement of a suitable alternative method to attain better accuracy in semiconducting and optoelectronic properties in different molecular devices. With this motivation, the “density of states (DOS) proportion” is proposed to incorporate a band and nonadiabatic hopping transport mechanism at different thermodynamic conditions. The DOS proportion is in direct proportion with the population of electronic states or normal DOS. In this context, the developed D/μ relation and other extended charge transport (CT) formalism (conductivity, current density, etc.) are used to study the CT in dialkyl substituted dithienothiophene (DSDTT)-based molecular crystals. The CT key parameters such as charge transfer integral, site energy, and spatial overlap integral are computed from the dimer projection (DIPRO) method using electronic structure calculations. These CT key parameters, including reorganization energy, are used in the degeneracy-weighted diffusion equation and the DOS proportion factor to explore the CT in the DSDTT-based molecular derivatives. Using the entropy-ruled method under degenerate and large carrier energy flux approximation, the calculated hole mobility values are around 0.85 and 0.19 cm2/V s for DSDTT-1 and DSDTT-2 molecular crystals, respectively, reaching a proximity value with an experimentally obtained average mobility. From the obtained ideality factor from the quantum-corrected Shockley diode equation, Langevin transport (trap-free diffusion) is observed in these DSDTT molecules, which suggests that these molecules are good candidates for designing hole transporting (or P-type semiconducting) devices.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


