{"title":"大规模基因组测序揭示了峡谷牦牛种群分离和选择特征。","authors":"Zemin Li, Jiahong Zhao, Xingyu Guo, Shiyu Wu, Shikai Wang, Jincheng Zhong, Yixi Kangzhu, Jikun Wang, Daoliang Lan, Jiabo Wang","doi":"10.1186/s12864-025-12114-7","DOIUrl":null,"url":null,"abstract":"<p><p>Through long-term natural and artificial selection, domestic yaks (Bos grunniens) have diverged from wild ancestors and become vital to high-altitude pastoralism, providing meat, milk, fiber, and other essential resources and transportation. Especially, based on the complex and changeable environments in the canyon of the Tibetan Plateau, the canyon-type yaks show diverse phenotypic traits, including morphology, production, and adaptation. To explore the genetic basis of this breed-specific adaptation and identify key functional genes shaped by selection, we collected 225 yaks from three canyon-type yak populations and scanned genome variation information using high-throughput resequencing. We employed three approaches, nucleotide diversity (π), fixation index (Fst) and cross-population extended haplotype homozygosity (XPEHH), to detect positive selection signals in the genome. Through analyses of population structure, genetic diversity, and selection sweep signals, we identified unique Single Nucleotide Polymorphisms (SNPs) that reveal significant genomic divergence among the three yak populations, which can also serve as genetic markers for population discrimination. Furthermore, shared SNPs identified by selective sweep analysis exhibited distinctive Minor Allele Frequency (MAF) distribution patterns; these population-specific SNP markers can be directly applied to develop SNP chips for breed-specific identification. A total of 13 genes related to breed-specific adaptive traits were identified. These findings provide valuable insights into the molecular signatures of breed-specific adaptation under human management and natural selection. It also identifies key functional genes relevant to future breeding programs.</p>","PeriodicalId":9030,"journal":{"name":"BMC Genomics","volume":"26 1","pages":"881"},"PeriodicalIF":3.7000,"publicationDate":"2025-10-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12495853/pdf/","citationCount":"0","resultStr":"{\"title\":\"Large-scale genome sequencing reveals population separation and selection signatures in major canyon yaks.\",\"authors\":\"Zemin Li, Jiahong Zhao, Xingyu Guo, Shiyu Wu, Shikai Wang, Jincheng Zhong, Yixi Kangzhu, Jikun Wang, Daoliang Lan, Jiabo Wang\",\"doi\":\"10.1186/s12864-025-12114-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Through long-term natural and artificial selection, domestic yaks (Bos grunniens) have diverged from wild ancestors and become vital to high-altitude pastoralism, providing meat, milk, fiber, and other essential resources and transportation. Especially, based on the complex and changeable environments in the canyon of the Tibetan Plateau, the canyon-type yaks show diverse phenotypic traits, including morphology, production, and adaptation. To explore the genetic basis of this breed-specific adaptation and identify key functional genes shaped by selection, we collected 225 yaks from three canyon-type yak populations and scanned genome variation information using high-throughput resequencing. We employed three approaches, nucleotide diversity (π), fixation index (Fst) and cross-population extended haplotype homozygosity (XPEHH), to detect positive selection signals in the genome. Through analyses of population structure, genetic diversity, and selection sweep signals, we identified unique Single Nucleotide Polymorphisms (SNPs) that reveal significant genomic divergence among the three yak populations, which can also serve as genetic markers for population discrimination. Furthermore, shared SNPs identified by selective sweep analysis exhibited distinctive Minor Allele Frequency (MAF) distribution patterns; these population-specific SNP markers can be directly applied to develop SNP chips for breed-specific identification. A total of 13 genes related to breed-specific adaptive traits were identified. These findings provide valuable insights into the molecular signatures of breed-specific adaptation under human management and natural selection. It also identifies key functional genes relevant to future breeding programs.</p>\",\"PeriodicalId\":9030,\"journal\":{\"name\":\"BMC Genomics\",\"volume\":\"26 1\",\"pages\":\"881\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2025-10-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12495853/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Genomics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12864-025-12114-7\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12864-025-12114-7","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
Large-scale genome sequencing reveals population separation and selection signatures in major canyon yaks.
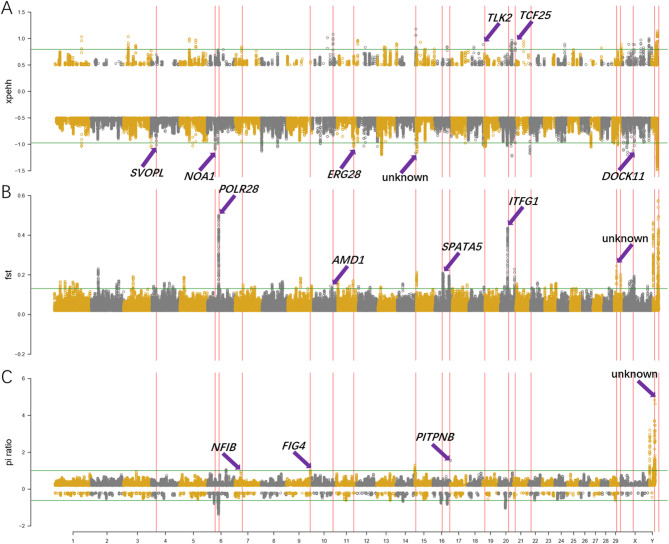
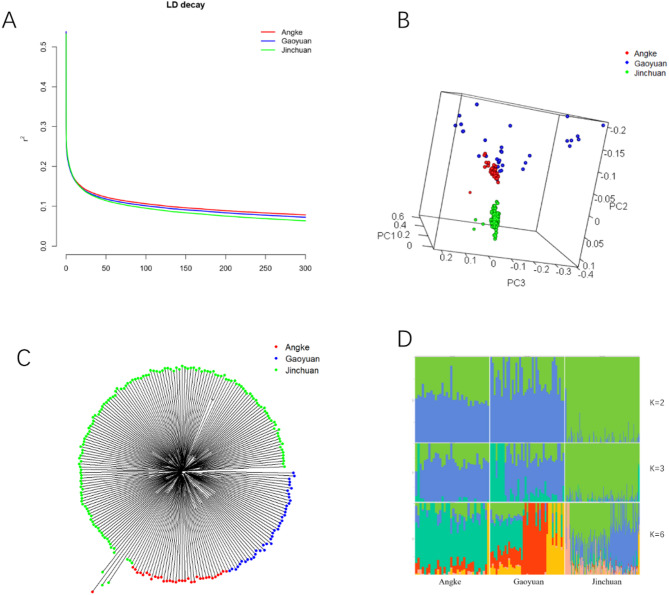
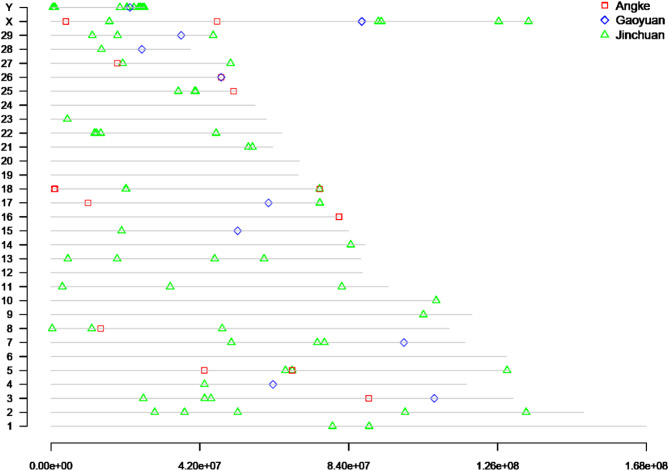
Through long-term natural and artificial selection, domestic yaks (Bos grunniens) have diverged from wild ancestors and become vital to high-altitude pastoralism, providing meat, milk, fiber, and other essential resources and transportation. Especially, based on the complex and changeable environments in the canyon of the Tibetan Plateau, the canyon-type yaks show diverse phenotypic traits, including morphology, production, and adaptation. To explore the genetic basis of this breed-specific adaptation and identify key functional genes shaped by selection, we collected 225 yaks from three canyon-type yak populations and scanned genome variation information using high-throughput resequencing. We employed three approaches, nucleotide diversity (π), fixation index (Fst) and cross-population extended haplotype homozygosity (XPEHH), to detect positive selection signals in the genome. Through analyses of population structure, genetic diversity, and selection sweep signals, we identified unique Single Nucleotide Polymorphisms (SNPs) that reveal significant genomic divergence among the three yak populations, which can also serve as genetic markers for population discrimination. Furthermore, shared SNPs identified by selective sweep analysis exhibited distinctive Minor Allele Frequency (MAF) distribution patterns; these population-specific SNP markers can be directly applied to develop SNP chips for breed-specific identification. A total of 13 genes related to breed-specific adaptive traits were identified. These findings provide valuable insights into the molecular signatures of breed-specific adaptation under human management and natural selection. It also identifies key functional genes relevant to future breeding programs.
期刊介绍:
BMC Genomics is an open access, peer-reviewed journal that considers articles on all aspects of genome-scale analysis, functional genomics, and proteomics.
BMC Genomics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


