Ni-P大块金属玻璃结构-成分-性能关系的研究
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
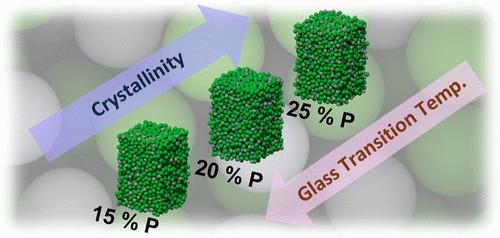
大块金属玻璃(bmg)是一类独特的材料,其特点是其无序的原子结构,赋予卓越的机械强度,耐腐蚀性和催化活性。然而,优化bmg的组成以获得所需的性能通常依赖于经验的试错,最多是由定性计算模型指导。在这里,我们将机器学习与电子结构理论相结合,定量绘制了ni -p基bmg的结构-组成-性质关系。我们使用基于神经网络的机器学习原子间势进行模拟,预测BMG的玻璃化转变温度随着磷含量的降低而降低,这与实验观察结果的定量一致。我们发现这种趋势与磷含量足够高时出现的材料的中等范围顺序相关。在原子尺度上,我们发现了以p为中心的簇基,它们的结构随组成和温度的变化而变化,并影响了Ni-P BMG中的原子迁移率。这种原子尺度的见解解释了Ni-P BMG的成分依赖稳定性,并展示了机器学习原子间势如何指导玻璃/非晶态材料(如BMG)的设计和优化。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Understanding Structure-Composition-Property Relationships of Ni-P Bulk Metallic Glasses
Bulk metallic glasses (BMGs) are a unique class of materials characterized by their disordered atomic structure, which imparts exceptional mechanical strength, corrosion resistance, and catalytic activity. Yet, optimizing the composition of BMGs for desired properties typically relies on empirical trial-and-error, at most guided by qualitative computational models. Here, we combined machine learning with electronic-structure theory to quantitatively map the structure-composition-property relationships of Ni–P-based BMGs. Our simulations using a neural-network-based machine learning interatomic potential predict that the glass transition temperature of the BMG decreases with the phosphorus content, in quantitative agreement with experimental observations. We find that this trend is correlated with medium-range order in the material that emerges when the phosphorus content is sufficiently high. On the atomic scale, we find P-centered cluster motifs that vary in structure with the composition and temperature and impact the atomic mobility in the Ni–P BMG. This atomic-scale insight explains the composition-dependent stability of the Ni–P BMG and demonstrates how machine-learning interatomic potentials can guide the design and optimization of glassy/amorphous materials such as BMGs.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


