Matthew W Foster, Youwei Chen, Marlene Violette, Michael T Forrester, J Scott Mellors, Brett S Phinney, Robert S Plumb, J Will Thompson, Timothy J McMahon
{"title":"开发和验证一种简化的工作流程,用于蛋白质组学分析和干燥血液的翻译后修饰。","authors":"Matthew W Foster, Youwei Chen, Marlene Violette, Michael T Forrester, J Scott Mellors, Brett S Phinney, Robert S Plumb, J Will Thompson, Timothy J McMahon","doi":"10.1101/2025.09.26.678912","DOIUrl":null,"url":null,"abstract":"<p><p>It is increasingly recognized that the 'omic analysis of whole blood has applications for precision medicine and disease phenotyping. Despite this realization, whole blood is generally viewed as a challenging analytical matrix in comparison to plasma or serum. Moreover, proteomic analyses of whole blood proteomics have almost exclusively focused on (non)targeted analyses of protein abundances and much less on post-translational modifications (PTMs). Here, we developed a streamlined workflow for processing twenty microliters of venous blood collected by volumetric absorptive microsampling that incorporates serial trypsinization, N-glycopeptide and phosphopeptide enrichment and avoids laborious sample dry-down or cleanup steps. Up to 10,000 analytes (reported as protein groups, glycopeptidoforms and phosphosites) were quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in approximately 2 h of MS acquisition time. Using these methods, we explored the stability of \"dried\" and \"wet\" blood proteomes, as well as effects of ex vivo inflammatory stimulus or phosphatase inhibition. Multi-omics factor analysis enabled facile identification of analytes that contributed to inter-individual variability of the blood proteomes, including N-glycopeptides that distinguish immunoglobulin heavy constant alpha 2 allotypes. Collectively, our results help to establish feasibility and best practices for the integrated MS-based quantification of proteins and PTMs from dried blood.</p>","PeriodicalId":519960,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12485729/pdf/","citationCount":"0","resultStr":"{\"title\":\"Development and validation of a streamlined workflow for proteomic analysis of proteins and post-translational modifications from dried blood.\",\"authors\":\"Matthew W Foster, Youwei Chen, Marlene Violette, Michael T Forrester, J Scott Mellors, Brett S Phinney, Robert S Plumb, J Will Thompson, Timothy J McMahon\",\"doi\":\"10.1101/2025.09.26.678912\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>It is increasingly recognized that the 'omic analysis of whole blood has applications for precision medicine and disease phenotyping. Despite this realization, whole blood is generally viewed as a challenging analytical matrix in comparison to plasma or serum. Moreover, proteomic analyses of whole blood proteomics have almost exclusively focused on (non)targeted analyses of protein abundances and much less on post-translational modifications (PTMs). Here, we developed a streamlined workflow for processing twenty microliters of venous blood collected by volumetric absorptive microsampling that incorporates serial trypsinization, N-glycopeptide and phosphopeptide enrichment and avoids laborious sample dry-down or cleanup steps. Up to 10,000 analytes (reported as protein groups, glycopeptidoforms and phosphosites) were quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in approximately 2 h of MS acquisition time. Using these methods, we explored the stability of \\\"dried\\\" and \\\"wet\\\" blood proteomes, as well as effects of ex vivo inflammatory stimulus or phosphatase inhibition. Multi-omics factor analysis enabled facile identification of analytes that contributed to inter-individual variability of the blood proteomes, including N-glycopeptides that distinguish immunoglobulin heavy constant alpha 2 allotypes. Collectively, our results help to establish feasibility and best practices for the integrated MS-based quantification of proteins and PTMs from dried blood.</p>\",\"PeriodicalId\":519960,\"journal\":{\"name\":\"bioRxiv : the preprint server for biology\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12485729/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"bioRxiv : the preprint server for biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/2025.09.26.678912\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2025.09.26.678912","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Development and validation of a streamlined workflow for proteomic analysis of proteins and post-translational modifications from dried blood.
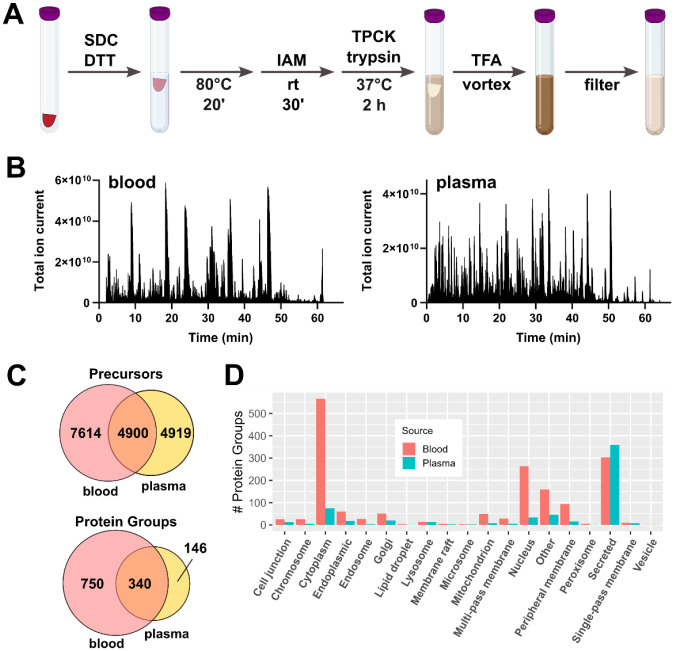
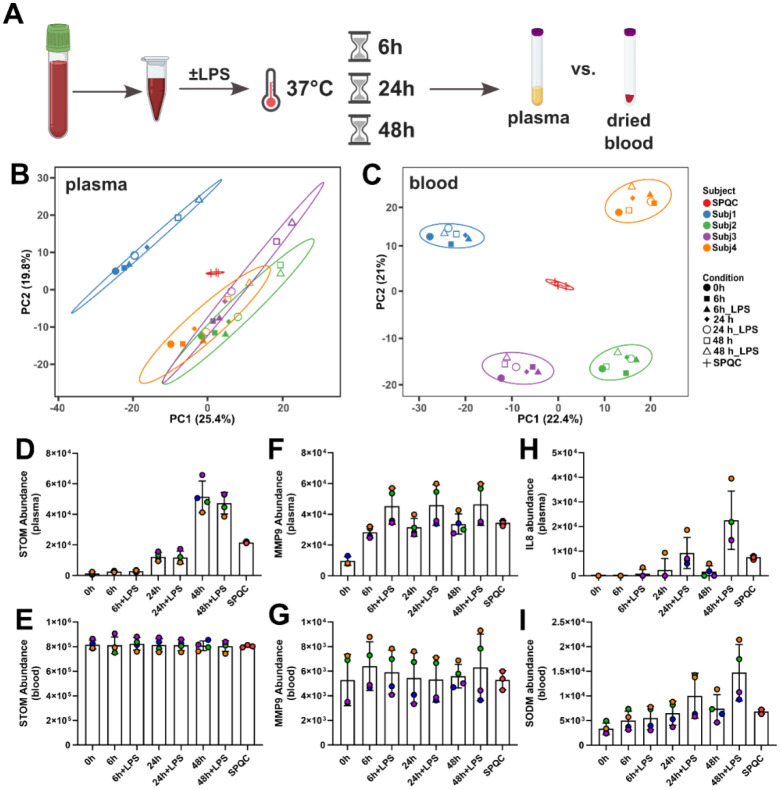
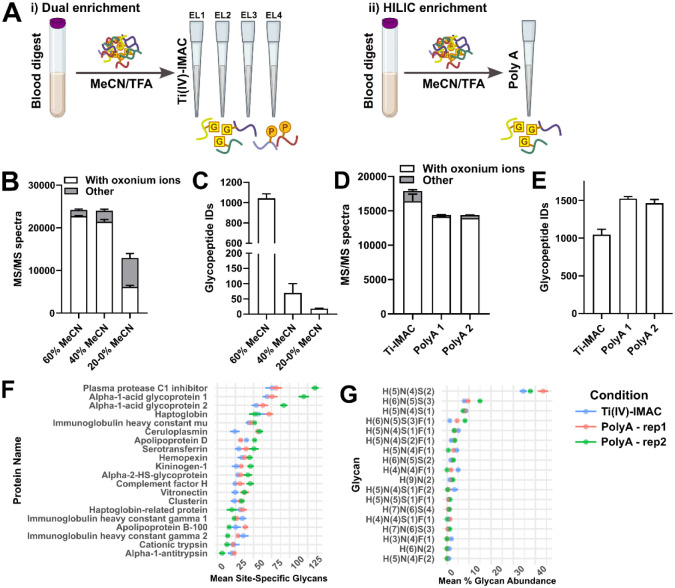
It is increasingly recognized that the 'omic analysis of whole blood has applications for precision medicine and disease phenotyping. Despite this realization, whole blood is generally viewed as a challenging analytical matrix in comparison to plasma or serum. Moreover, proteomic analyses of whole blood proteomics have almost exclusively focused on (non)targeted analyses of protein abundances and much less on post-translational modifications (PTMs). Here, we developed a streamlined workflow for processing twenty microliters of venous blood collected by volumetric absorptive microsampling that incorporates serial trypsinization, N-glycopeptide and phosphopeptide enrichment and avoids laborious sample dry-down or cleanup steps. Up to 10,000 analytes (reported as protein groups, glycopeptidoforms and phosphosites) were quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in approximately 2 h of MS acquisition time. Using these methods, we explored the stability of "dried" and "wet" blood proteomes, as well as effects of ex vivo inflammatory stimulus or phosphatase inhibition. Multi-omics factor analysis enabled facile identification of analytes that contributed to inter-individual variability of the blood proteomes, including N-glycopeptides that distinguish immunoglobulin heavy constant alpha 2 allotypes. Collectively, our results help to establish feasibility and best practices for the integrated MS-based quantification of proteins and PTMs from dried blood.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


