{"title":"抗癌类黄酮作为登革热病毒复制抑制剂的硅鉴定:分子对接和模拟方法","authors":"Soumendu Patra, Arindam Paul, Harshita Shand, Sayan Ghosal, Suvankar Ghorai","doi":"10.1007/s00894-025-06516-3","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Dengue, a mosquito-borne viral disease endemic to over 100 countries, poses a serious health risk to people living in tropical regions. The viral non-structural proteins NS3 (helicase) and NS5 (RNA-dependent RNA polymerase) are critical targets for antiviral drug development. Several natural and synthetic compounds have been tried against this for the screening of antiviral inhibitor(s) but so far limited success has been achieved. In this study, we have investigated how natural products interact with and destabilize NS3 and NS5. We used in silico methods to screen new potential NS3 and NS5 inhibitors from various anti-cancer flavonoid compounds that previously showed anti-cancer properties in vitro. A virtual screening was conducted on 329 anti-cancer flavonoid compounds, selecting 190 compounds based on Lipinski’s rule of five, the Muegge filter, the Ghose filter, and the Veber filter. Molecular docking techniques allowed us to identify Artobiloxanthone (PubChem CID: 46887866) as the most effective binder for NS3, while Glabridin (PubChem CID: 124052) emerged as the most effective binder for NS5. These leading candidates demonstrated favorable ADMET profiles. Additionally, molecular dynamics (MD) simulations of up to 1000-ns showcased stable protein–ligand interactions, with convergence achieved at 200 ns for NS3 and 470 ns for NS5. The structural stability of the complexes was validated by analyzing root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), and hydrogen bonding (H-bonding). Binding affinity between the ligand and target proteins was further validated by binding free energy calculations using the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) approach, providing a robust estimation of interaction stability. These findings highlight the potential of Artobiloxanthone and Glabridin as promising inhibitors of dengue virus replication.</p><h3>Methods</h3><p>The Chimera v1.11.2 program was employed for protein optimization, while PyRx 0.8 served for molecular docking. Protein structures were converted into.pdbqt format, and ligands were energy-minimized using the MMFF94 force field and conjugate gradient optimization. Visualization was conducted using BIOVIA Discovery Studio Visualizer and PyMOL. The ADMET properties of the top hits were predicted using the pkCSM and ProTox-II platforms. To address missing residues in the crystal structures, AlphaFold2 was used to predict full-length protein models. MD simulations were performed with the GROMACS 2024.5 package, utilizing the AMBER-f99SB-ILDN force field, the TIP3P water model, and a 120 mM NaCl concentration. Equilibration was achieved through V-rescale and C-rescale thermostats, followed by Parrinello–Rahman barostat production runs. Analysis of the MD trajectories included RMSD, RMSF, Rg, SASA, H-bonding, and MM-PBSA utilizing GROMACS tools, gmx_MMPBSA, VMD, and MDAnalysis.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 11","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"In silico identification of anticancer flavonoids as dengue virus replication inhibitors: a molecular docking and simulation approach\",\"authors\":\"Soumendu Patra, Arindam Paul, Harshita Shand, Sayan Ghosal, Suvankar Ghorai\",\"doi\":\"10.1007/s00894-025-06516-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>Dengue, a mosquito-borne viral disease endemic to over 100 countries, poses a serious health risk to people living in tropical regions. The viral non-structural proteins NS3 (helicase) and NS5 (RNA-dependent RNA polymerase) are critical targets for antiviral drug development. Several natural and synthetic compounds have been tried against this for the screening of antiviral inhibitor(s) but so far limited success has been achieved. In this study, we have investigated how natural products interact with and destabilize NS3 and NS5. We used in silico methods to screen new potential NS3 and NS5 inhibitors from various anti-cancer flavonoid compounds that previously showed anti-cancer properties in vitro. A virtual screening was conducted on 329 anti-cancer flavonoid compounds, selecting 190 compounds based on Lipinski’s rule of five, the Muegge filter, the Ghose filter, and the Veber filter. Molecular docking techniques allowed us to identify Artobiloxanthone (PubChem CID: 46887866) as the most effective binder for NS3, while Glabridin (PubChem CID: 124052) emerged as the most effective binder for NS5. These leading candidates demonstrated favorable ADMET profiles. Additionally, molecular dynamics (MD) simulations of up to 1000-ns showcased stable protein–ligand interactions, with convergence achieved at 200 ns for NS3 and 470 ns for NS5. The structural stability of the complexes was validated by analyzing root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), and hydrogen bonding (H-bonding). Binding affinity between the ligand and target proteins was further validated by binding free energy calculations using the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) approach, providing a robust estimation of interaction stability. These findings highlight the potential of Artobiloxanthone and Glabridin as promising inhibitors of dengue virus replication.</p><h3>Methods</h3><p>The Chimera v1.11.2 program was employed for protein optimization, while PyRx 0.8 served for molecular docking. Protein structures were converted into.pdbqt format, and ligands were energy-minimized using the MMFF94 force field and conjugate gradient optimization. Visualization was conducted using BIOVIA Discovery Studio Visualizer and PyMOL. The ADMET properties of the top hits were predicted using the pkCSM and ProTox-II platforms. To address missing residues in the crystal structures, AlphaFold2 was used to predict full-length protein models. MD simulations were performed with the GROMACS 2024.5 package, utilizing the AMBER-f99SB-ILDN force field, the TIP3P water model, and a 120 mM NaCl concentration. Equilibration was achieved through V-rescale and C-rescale thermostats, followed by Parrinello–Rahman barostat production runs. Analysis of the MD trajectories included RMSD, RMSF, Rg, SASA, H-bonding, and MM-PBSA utilizing GROMACS tools, gmx_MMPBSA, VMD, and MDAnalysis.</p><h3>Graphical Abstract</h3>\\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"31 11\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-10-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-025-06516-3\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06516-3","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
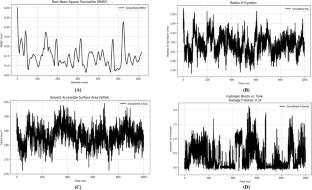
登革热是一种蚊子传播的病毒性疾病,在100多个国家流行,对生活在热带地区的人们构成严重的健康风险。病毒非结构蛋白NS3(解旋酶)和NS5 (RNA依赖性RNA聚合酶)是抗病毒药物开发的重要靶点。已经尝试了几种天然和合成化合物来筛选抗病毒抑制剂,但迄今为止取得的成功有限。在本研究中,我们研究了天然产物如何与NS3和NS5相互作用并使其失稳。我们利用计算机方法从各种抗癌类黄酮化合物中筛选新的潜在的NS3和NS5抑制剂,这些化合物先前在体外显示出抗癌特性。对329种抗癌类黄酮化合物进行虚拟筛选,根据Lipinski的五法则、Muegge过滤器、Ghose过滤器和Veber过滤器筛选出190种化合物。分子对接技术使我们确定Artobiloxanthone (PubChem CID: 46887866)是NS3最有效的结合剂,而光定(PubChem CID: 124052)是NS5最有效的结合剂。这些领先的候选人表现出良好的ADMET概况。此外,分子动力学(MD)模拟显示,高达1000-ns的蛋白质-配体相互作用稳定,NS3和NS5分别在200 ns和470 ns下实现收敛。通过分析配合物的均方根偏差(RMSD)、均方根波动(RMSF)、旋转半径(Rg)、溶剂可及表面积(SASA)和氢键(h -键)来验证配合物的结构稳定性。利用分子力学泊松-玻尔兹曼表面积(MM-PBSA)方法计算结合自由能,进一步验证了配体与靶蛋白之间的结合亲和力,提供了对相互作用稳定性的可靠估计。这些发现突出了Artobiloxanthone和光定作为登革热病毒复制抑制剂的潜力。方法采用Chimera v1.11.2程序进行蛋白优化,PyRx 0.8程序进行分子对接。蛋白质结构被转化为。采用MMFF94力场和共轭梯度优化对配体进行能量最小化。使用BIOVIA Discovery Studio Visualizer和PyMOL进行可视化。利用pkCSM和ProTox-II平台预测了顶击的ADMET特性。为了解决晶体结构中缺失的残基,使用AlphaFold2来预测全长蛋白质模型。采用GROMACS 2024.5软件包,利用AMBER-f99SB-ILDN力场、TIP3P水模型和120 mM NaCl浓度进行MD模拟。平衡是通过V-rescale和C-rescale恒温器实现的,然后是Parrinello-Rahman恒压器生产运行。MD轨迹分析包括RMSD、RMSF、Rg、SASA、h键和MM-PBSA,使用GROMACS工具,gmx_MMPBSA、VMD和MDAnalysis。图形抽象
In silico identification of anticancer flavonoids as dengue virus replication inhibitors: a molecular docking and simulation approach
Context
Dengue, a mosquito-borne viral disease endemic to over 100 countries, poses a serious health risk to people living in tropical regions. The viral non-structural proteins NS3 (helicase) and NS5 (RNA-dependent RNA polymerase) are critical targets for antiviral drug development. Several natural and synthetic compounds have been tried against this for the screening of antiviral inhibitor(s) but so far limited success has been achieved. In this study, we have investigated how natural products interact with and destabilize NS3 and NS5. We used in silico methods to screen new potential NS3 and NS5 inhibitors from various anti-cancer flavonoid compounds that previously showed anti-cancer properties in vitro. A virtual screening was conducted on 329 anti-cancer flavonoid compounds, selecting 190 compounds based on Lipinski’s rule of five, the Muegge filter, the Ghose filter, and the Veber filter. Molecular docking techniques allowed us to identify Artobiloxanthone (PubChem CID: 46887866) as the most effective binder for NS3, while Glabridin (PubChem CID: 124052) emerged as the most effective binder for NS5. These leading candidates demonstrated favorable ADMET profiles. Additionally, molecular dynamics (MD) simulations of up to 1000-ns showcased stable protein–ligand interactions, with convergence achieved at 200 ns for NS3 and 470 ns for NS5. The structural stability of the complexes was validated by analyzing root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), and hydrogen bonding (H-bonding). Binding affinity between the ligand and target proteins was further validated by binding free energy calculations using the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) approach, providing a robust estimation of interaction stability. These findings highlight the potential of Artobiloxanthone and Glabridin as promising inhibitors of dengue virus replication.
Methods
The Chimera v1.11.2 program was employed for protein optimization, while PyRx 0.8 served for molecular docking. Protein structures were converted into.pdbqt format, and ligands were energy-minimized using the MMFF94 force field and conjugate gradient optimization. Visualization was conducted using BIOVIA Discovery Studio Visualizer and PyMOL. The ADMET properties of the top hits were predicted using the pkCSM and ProTox-II platforms. To address missing residues in the crystal structures, AlphaFold2 was used to predict full-length protein models. MD simulations were performed with the GROMACS 2024.5 package, utilizing the AMBER-f99SB-ILDN force field, the TIP3P water model, and a 120 mM NaCl concentration. Equilibration was achieved through V-rescale and C-rescale thermostats, followed by Parrinello–Rahman barostat production runs. Analysis of the MD trajectories included RMSD, RMSF, Rg, SASA, H-bonding, and MM-PBSA utilizing GROMACS tools, gmx_MMPBSA, VMD, and MDAnalysis.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


