相关量热法和电位滴定法阐明了锂离子配位热力学的定量
IF 15.6
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
了解锂离子配位环境中相互作用的热力学对合理的电解质设计至关重要。在这项工作中,我们与生物系统中的配体-受体结合进行了物理类比,引入了一种结合等温滴定量热法(ITC)和电位滴定法(PT)的框架,以定量确定与溶剂位移相关的吉布斯自由能(ΔG)、焓(ΔH)和熵(ΔS),这是一种典型的配位球反应,有力地报告了非水锂基电解质中潜在的能量学。首先用拉曼光谱验证了溶剂置换方案背后的物理理解,这样,在ITC期间测量的热量被证实主要是由配合溶剂的差异变化引起的,因为大块电解质成分被系统地修改了。然后建立了一个统计结合模型来解释热力学数据,并参数化了一组典型双溶剂电解质的单位位移焓和平衡常数。该框架在不同溶剂(DMSO:乙腈或DMSO:PC)和结构相似溶剂(EC:PC)的体系中进行了测试,揭示了溶剂-阳离子相互作用焓和熵的细微差异如何控制配位偏好。这些结果为微观配位球变化背后的驱动力提供了新的实验见解,并为指导电解质配方提供了有力的方法。本文章由计算机程序翻译,如有差异,请以英文原文为准。
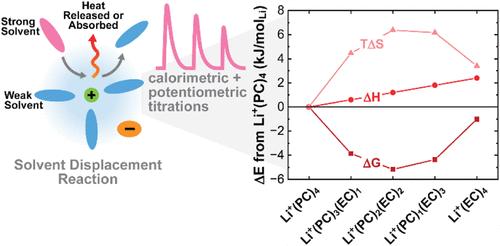
Correlated Calorimetric and Potentiometric Titration Elucidates Quantitative Lithium Cation Coordination Thermodynamics
Understanding the thermodynamics of interactions within the coordination environment of Li+ ions is critical for rational electrolyte design. Drawing a physical analogy to ligand–receptor binding in biological systems, in this work, we introduce a combined isothermal titration calorimetry (ITC) and potentiometric titration (PT) framework to quantitatively determine the Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (ΔS) associated with solvent displacement─an exemplar coordination sphere reaction that powerfully reports on the underlying energetics─in nonaqueous Li-based electrolytes. The physical understanding behind the solvent displacement scheme is first validated with Raman spectroscopy, such that the heat measured during ITC is confirmed to arise predominantly from differential changes in coordinated solvent as the bulk electrolyte composition is systematically modified. A statistical binding model is then developed to interpret the thermodynamic data and parametrize single-site displacement enthalpy and equilibrium constants in a set of exemplar dual-solvent electrolytes. The framework is tested across systems with both dissimilar (DMSO:acetonitrile or DMSO:PC) and structurally similar (EC:PC) solvents, revealing how subtle differences in solvent–cation interaction enthalpy and entropy govern coordination preferences. These results provide new experimental insight into the driving forces behind microscopic coordination sphere changes and offer a powerful approach to guide electrolyte formulation.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


