{"title":"发现系统发育网络之间最大的共同收缩。","authors":"Bertrand Marchand, Nadia Tahiri, Shohreh Golpaigani Fard, Olivier Tremblay-Savard, Manuel Lafond","doi":"10.1186/s13015-025-00283-9","DOIUrl":null,"url":null,"abstract":"<p><p>In this paper, we lay the groundwork on the comparison of phylogenetic networks based on edge contractions and expansions as edit operations, as originally proposed by Robinson and Foulds to compare trees. We prove that these operations connect the space of all phylogenetic networks on the same set of leaves, even if we forbid contractions that create cycles. This allows to define an operational distance on this space, as the minimum number of contractions and expansions required to transform one network into another. We highlight the difference between this distance and the computation of the maximum common contraction between two networks. Given its ability to outline a common structure between them, which can provide valuable biological insights, we study the algorithmic aspects of the latter. We first prove that computing a maximum common contraction between two networks is NP-hard, even when the maximum degree, the size of the common contraction, or the number of leaves is bounded. We also provide lower bounds to the problem based on the Exponential-Time Hypothesis. Nonetheless, we do provide a polynomial-time algorithm for weakly galled trees, a generalization of galled trees.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"20 1","pages":"18"},"PeriodicalIF":1.7000,"publicationDate":"2025-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12490124/pdf/","citationCount":"0","resultStr":"{\"title\":\"Finding maximum common contractions between phylogenetic networks.\",\"authors\":\"Bertrand Marchand, Nadia Tahiri, Shohreh Golpaigani Fard, Olivier Tremblay-Savard, Manuel Lafond\",\"doi\":\"10.1186/s13015-025-00283-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>In this paper, we lay the groundwork on the comparison of phylogenetic networks based on edge contractions and expansions as edit operations, as originally proposed by Robinson and Foulds to compare trees. We prove that these operations connect the space of all phylogenetic networks on the same set of leaves, even if we forbid contractions that create cycles. This allows to define an operational distance on this space, as the minimum number of contractions and expansions required to transform one network into another. We highlight the difference between this distance and the computation of the maximum common contraction between two networks. Given its ability to outline a common structure between them, which can provide valuable biological insights, we study the algorithmic aspects of the latter. We first prove that computing a maximum common contraction between two networks is NP-hard, even when the maximum degree, the size of the common contraction, or the number of leaves is bounded. We also provide lower bounds to the problem based on the Exponential-Time Hypothesis. Nonetheless, we do provide a polynomial-time algorithm for weakly galled trees, a generalization of galled trees.</p>\",\"PeriodicalId\":50823,\"journal\":{\"name\":\"Algorithms for Molecular Biology\",\"volume\":\"20 1\",\"pages\":\"18\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12490124/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Algorithms for Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13015-025-00283-9\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-025-00283-9","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Finding maximum common contractions between phylogenetic networks.
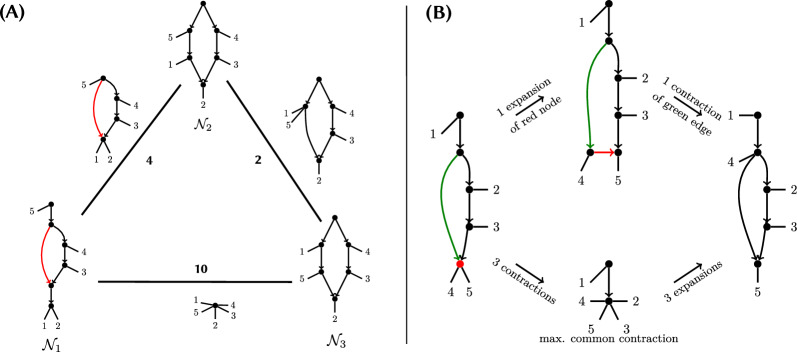
In this paper, we lay the groundwork on the comparison of phylogenetic networks based on edge contractions and expansions as edit operations, as originally proposed by Robinson and Foulds to compare trees. We prove that these operations connect the space of all phylogenetic networks on the same set of leaves, even if we forbid contractions that create cycles. This allows to define an operational distance on this space, as the minimum number of contractions and expansions required to transform one network into another. We highlight the difference between this distance and the computation of the maximum common contraction between two networks. Given its ability to outline a common structure between them, which can provide valuable biological insights, we study the algorithmic aspects of the latter. We first prove that computing a maximum common contraction between two networks is NP-hard, even when the maximum degree, the size of the common contraction, or the number of leaves is bounded. We also provide lower bounds to the problem based on the Exponential-Time Hypothesis. Nonetheless, we do provide a polynomial-time algorithm for weakly galled trees, a generalization of galled trees.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


