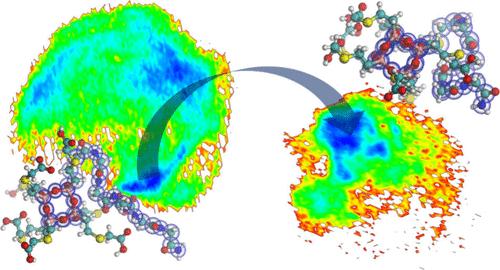
{"title":"poss -肽共轭分子在选择性溶剂中的可调构象性质:原子分子动力学模拟研究。","authors":"Junhao Dai, , , Wen Tang, , , Xianbo Huang*, , and , Rui Zhang*, ","doi":"10.1021/acs.jpcb.5c05243","DOIUrl":null,"url":null,"abstract":"<p >Giant molecules with precisely defined modular architectures hold promise to generate distinct structure-dynamics-property relationships in solution-phase materials. Recently, a novel class of hybrid macromolecules that combine structural rigidity of polyhedral oligomeric silsesquioxanes (POSS) with flexibility of peptide sequences (POSS-peptide conjugate molecules) have been established experimentally. To elucidate their detailed microscopic structural and dynamic features, high-precision atomistic modeling and simulation are in demand. In this study, we develop a standardized and extensible all-atom force field parametrization workflow for POSS-peptide molecules, integrating quantum chemical calculations to derive accurate force field parameters for the rigid POSS units, including bond, angle, and dihedral terms, as well as atomic charges for the whole molecule. Upon applying the parametrization framework to five representative POSS-peptide molecules with varied POSS functionality and peptide composition in water or DMF solvent, all-atom molecular dynamics simulations are performed for the ten systems to investigate the highly tunable conformational properties of POSS-peptides. We construct detailed conformational free energy landscapes that provide insight into the role of different factors in shaping the molecule’s solution-phase behavior. Our analysis reveals that molecular structure and solvent polarity co-regulate the conformational preferences of POSS-peptide molecules. Of particular interest is the finding of some unusual structure-dynamics correlation behaviors driven by close-distance interactions between the POSS and peptide unit. This work expands our understanding of the conformational richness of POSS-peptides in solution and provides a methodology foundation for exploring larger-scale supramolecular structures achievable by this emergent family of giant molecules in future research.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"129 41","pages":"10903–10913"},"PeriodicalIF":2.9000,"publicationDate":"2025-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Tunable Conformational Properties of POSS-Peptide Conjugate Molecule in Selective Solvents: An Atomistic Molecular Dynamics Simulation Study\",\"authors\":\"Junhao Dai, , , Wen Tang, , , Xianbo Huang*, , and , Rui Zhang*, \",\"doi\":\"10.1021/acs.jpcb.5c05243\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Giant molecules with precisely defined modular architectures hold promise to generate distinct structure-dynamics-property relationships in solution-phase materials. Recently, a novel class of hybrid macromolecules that combine structural rigidity of polyhedral oligomeric silsesquioxanes (POSS) with flexibility of peptide sequences (POSS-peptide conjugate molecules) have been established experimentally. To elucidate their detailed microscopic structural and dynamic features, high-precision atomistic modeling and simulation are in demand. In this study, we develop a standardized and extensible all-atom force field parametrization workflow for POSS-peptide molecules, integrating quantum chemical calculations to derive accurate force field parameters for the rigid POSS units, including bond, angle, and dihedral terms, as well as atomic charges for the whole molecule. Upon applying the parametrization framework to five representative POSS-peptide molecules with varied POSS functionality and peptide composition in water or DMF solvent, all-atom molecular dynamics simulations are performed for the ten systems to investigate the highly tunable conformational properties of POSS-peptides. We construct detailed conformational free energy landscapes that provide insight into the role of different factors in shaping the molecule’s solution-phase behavior. Our analysis reveals that molecular structure and solvent polarity co-regulate the conformational preferences of POSS-peptide molecules. Of particular interest is the finding of some unusual structure-dynamics correlation behaviors driven by close-distance interactions between the POSS and peptide unit. This work expands our understanding of the conformational richness of POSS-peptides in solution and provides a methodology foundation for exploring larger-scale supramolecular structures achievable by this emergent family of giant molecules in future research.</p>\",\"PeriodicalId\":60,\"journal\":{\"name\":\"The Journal of Physical Chemistry B\",\"volume\":\"129 41\",\"pages\":\"10903–10913\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry B\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcb.5c05243\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.5c05243","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Tunable Conformational Properties of POSS-Peptide Conjugate Molecule in Selective Solvents: An Atomistic Molecular Dynamics Simulation Study
Giant molecules with precisely defined modular architectures hold promise to generate distinct structure-dynamics-property relationships in solution-phase materials. Recently, a novel class of hybrid macromolecules that combine structural rigidity of polyhedral oligomeric silsesquioxanes (POSS) with flexibility of peptide sequences (POSS-peptide conjugate molecules) have been established experimentally. To elucidate their detailed microscopic structural and dynamic features, high-precision atomistic modeling and simulation are in demand. In this study, we develop a standardized and extensible all-atom force field parametrization workflow for POSS-peptide molecules, integrating quantum chemical calculations to derive accurate force field parameters for the rigid POSS units, including bond, angle, and dihedral terms, as well as atomic charges for the whole molecule. Upon applying the parametrization framework to five representative POSS-peptide molecules with varied POSS functionality and peptide composition in water or DMF solvent, all-atom molecular dynamics simulations are performed for the ten systems to investigate the highly tunable conformational properties of POSS-peptides. We construct detailed conformational free energy landscapes that provide insight into the role of different factors in shaping the molecule’s solution-phase behavior. Our analysis reveals that molecular structure and solvent polarity co-regulate the conformational preferences of POSS-peptide molecules. Of particular interest is the finding of some unusual structure-dynamics correlation behaviors driven by close-distance interactions between the POSS and peptide unit. This work expands our understanding of the conformational richness of POSS-peptides in solution and provides a methodology foundation for exploring larger-scale supramolecular structures achievable by this emergent family of giant molecules in future research.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


