独家功能签名基因注释与广阔的OpenOrd布局。
IF 1.7
4区 生物学
Q4 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
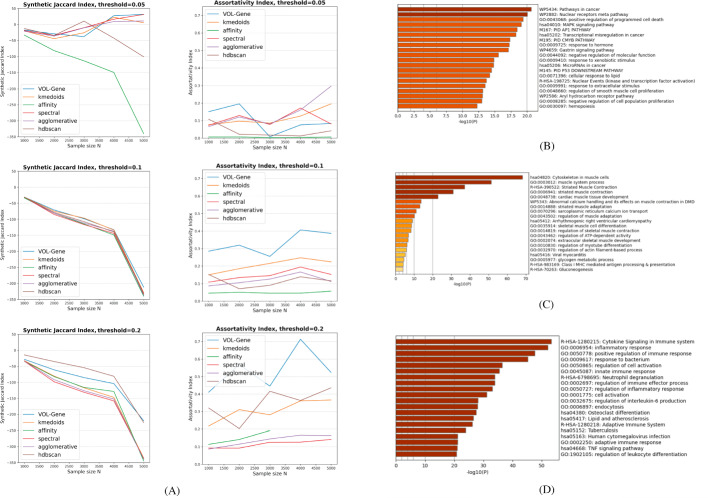
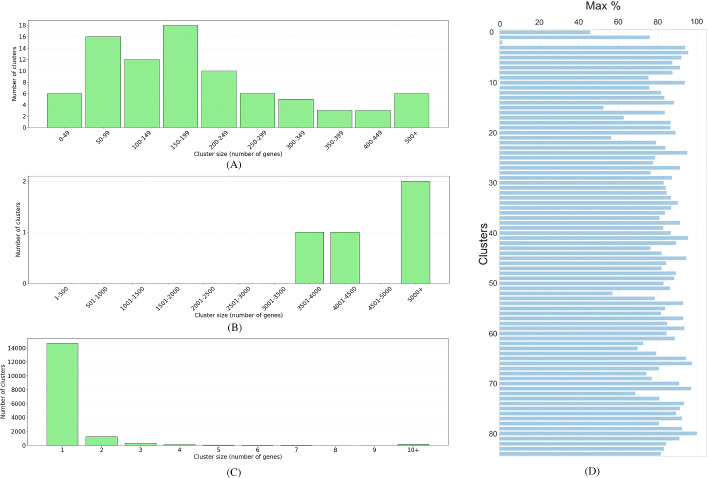
生物学研究可以产生有限数量的标记基因,不足以用于基因集富集分析。在这里,我们提出了volg - gene,这是一种基于图的算法,它将所有基因划分为功能相关基因的非重叠类,从而为每个基因分配单个功能。为此,将许多功能签名组合成一个加权图,并将其划分为团。对于一个标记基因,我们的方法获取了许多属于同一类的基因,其中一些基因可以被很好地注释,并且可能参与相同的生物过程。本文章由计算机程序翻译,如有差异,请以英文原文为准。

Exclusive functional signatures for gene annotation with vast OpenOrd layout.
A biological study can produce a limited number of marker genes, not large enough to be used in gene set enrichment analysis. Here we suggest VOL-Gene, a graph-based algorithm that partitions all genes into non-overlapping classes of functionally related genes, thus assigning a single function to each gene. To this end, many functional signatures are combined into a single weighted graph, which is partitioned into cliques. For a poorly annotated marker gene, our approach fetches a number of genes that belong to the same class, some of which can be well annotated and are likely to take part in the same biological process.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Algorithms for Molecular Biology
生物-生化研究方法
CiteScore
2.40
自引率
10.00%
发文量
16
审稿时长
>12 weeks
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


