Alessia Giovanna Andrisano, Nadia Castaldo, Francesco Giuliana, Davide Femia, Giuseppe Morana, Vincenzo Patruno, Giorgio Monteleone, Nicolò Reccardini, Rossella Cifaldi, Michael Hughes, Yukai Wang, Paola Confalonieri, Francesco Salton, Pietro Geri, Marco Confalonieri, Barbara Ruaro
{"title":"尼达尼布治疗特发性和进行性肺纤维化的回顾性观察研究。","authors":"Alessia Giovanna Andrisano, Nadia Castaldo, Francesco Giuliana, Davide Femia, Giuseppe Morana, Vincenzo Patruno, Giorgio Monteleone, Nicolò Reccardini, Rossella Cifaldi, Michael Hughes, Yukai Wang, Paola Confalonieri, Francesco Salton, Pietro Geri, Marco Confalonieri, Barbara Ruaro","doi":"10.3390/jcm14186665","DOIUrl":null,"url":null,"abstract":"<p><p><b>Background/Objectives:</b> Idiopathic pulmonary fibrosis (IPF) is the most common form of pulmonary fibrosis (PF) and serves as a key reference for disease severity. Progressive pulmonary fibrosis (PPF), a distinct yet heterogeneous entity arising from various interstitial lung diseases (ILDs), shares similar pathogenetic mechanisms and clinical courses driven by self-perpetuating fibrosis. Antifibrotic therapy, notably nintedanib, can slow disease progression. However, real-world data on antifibrotic therapy's impact on survival, especially in PPF, are limited. This study aims to compare IPF and PPF regarding phenotype, radiological patterns, comorbidities, prognostic factors, and response to nintedanib, focusing on identifying the patient subsets most likely to benefit. Outcomes assessed include safety, survival, and disease progression over one year, considering various prognostic factors. <b>Methods:</b> This retrospective observational study evaluated patients with fibrosing ILD, affected by either IPF or PPF, and treated with nintedanib. Data collected encompassed clinical, radiological, functional, and treatment-related information. Assessments included chest CT, pulmonary function tests, comorbidities, and survival analysis, utilizing standardized methods and statistical tools to interpret outcomes and tolerability. <b>Results:</b> The study population was composed of 97 patients: 64 were diagnosed with IPF and 33 with PPF. The analysis showed that in PPF patients, ongoing antifibrotic treatment resulted in higher survival (71.1 months vs. 27.4 months, <i>p</i> < 0.001), while no statistically significant differences were found in the IPF group (67.4 months vs. 52.5 months, <i>p</i> = 0.216). Nintedanib was generally well tolerated. Gastrointestinal side effects, predominantly diarrhea, were reported in 61% of patients with IPF and 50% of those with PPF. Dose reduction occurred in 43.75% of IPF patients and 36% of PPF patients, while treatment discontinuation was required in 21.87% of IPF and 21% of PPF patients. <b>Conclusions:</b> This study highlights that in PPF patients, antifibrotic therapy with nintedanib can improve survival. This statement underlines that the primary outcome of antifibrotic treatment should focus on improving patients' survival.</p>","PeriodicalId":15533,"journal":{"name":"Journal of Clinical Medicine","volume":"14 18","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2025-09-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12471148/pdf/","citationCount":"0","resultStr":"{\"title\":\"Retrospective Observational Study of Nintedanib in Managing Idiopathic and Progressive Pulmonary Fibrosis in Routine Practice.\",\"authors\":\"Alessia Giovanna Andrisano, Nadia Castaldo, Francesco Giuliana, Davide Femia, Giuseppe Morana, Vincenzo Patruno, Giorgio Monteleone, Nicolò Reccardini, Rossella Cifaldi, Michael Hughes, Yukai Wang, Paola Confalonieri, Francesco Salton, Pietro Geri, Marco Confalonieri, Barbara Ruaro\",\"doi\":\"10.3390/jcm14186665\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p><b>Background/Objectives:</b> Idiopathic pulmonary fibrosis (IPF) is the most common form of pulmonary fibrosis (PF) and serves as a key reference for disease severity. Progressive pulmonary fibrosis (PPF), a distinct yet heterogeneous entity arising from various interstitial lung diseases (ILDs), shares similar pathogenetic mechanisms and clinical courses driven by self-perpetuating fibrosis. Antifibrotic therapy, notably nintedanib, can slow disease progression. However, real-world data on antifibrotic therapy's impact on survival, especially in PPF, are limited. This study aims to compare IPF and PPF regarding phenotype, radiological patterns, comorbidities, prognostic factors, and response to nintedanib, focusing on identifying the patient subsets most likely to benefit. Outcomes assessed include safety, survival, and disease progression over one year, considering various prognostic factors. <b>Methods:</b> This retrospective observational study evaluated patients with fibrosing ILD, affected by either IPF or PPF, and treated with nintedanib. Data collected encompassed clinical, radiological, functional, and treatment-related information. Assessments included chest CT, pulmonary function tests, comorbidities, and survival analysis, utilizing standardized methods and statistical tools to interpret outcomes and tolerability. <b>Results:</b> The study population was composed of 97 patients: 64 were diagnosed with IPF and 33 with PPF. The analysis showed that in PPF patients, ongoing antifibrotic treatment resulted in higher survival (71.1 months vs. 27.4 months, <i>p</i> < 0.001), while no statistically significant differences were found in the IPF group (67.4 months vs. 52.5 months, <i>p</i> = 0.216). Nintedanib was generally well tolerated. Gastrointestinal side effects, predominantly diarrhea, were reported in 61% of patients with IPF and 50% of those with PPF. Dose reduction occurred in 43.75% of IPF patients and 36% of PPF patients, while treatment discontinuation was required in 21.87% of IPF and 21% of PPF patients. <b>Conclusions:</b> This study highlights that in PPF patients, antifibrotic therapy with nintedanib can improve survival. This statement underlines that the primary outcome of antifibrotic treatment should focus on improving patients' survival.</p>\",\"PeriodicalId\":15533,\"journal\":{\"name\":\"Journal of Clinical Medicine\",\"volume\":\"14 18\",\"pages\":\"\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-09-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12471148/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.3390/jcm14186665\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3390/jcm14186665","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
Retrospective Observational Study of Nintedanib in Managing Idiopathic and Progressive Pulmonary Fibrosis in Routine Practice.
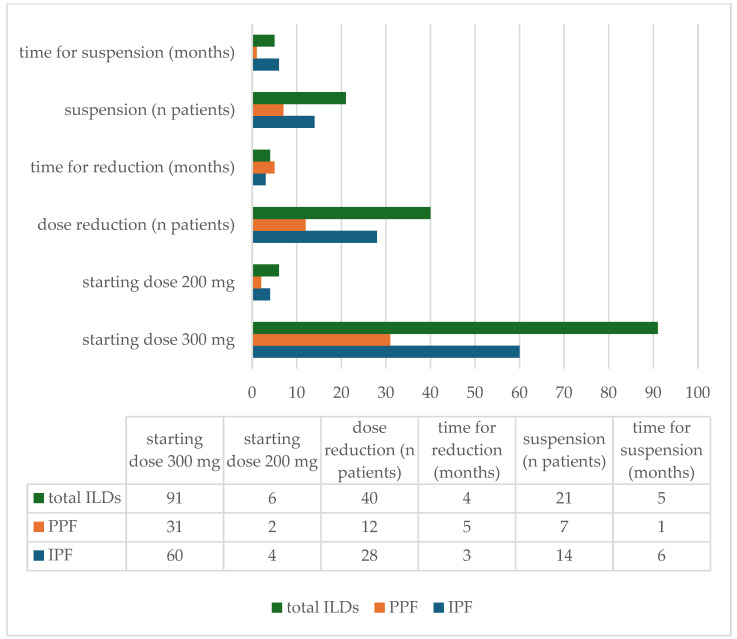
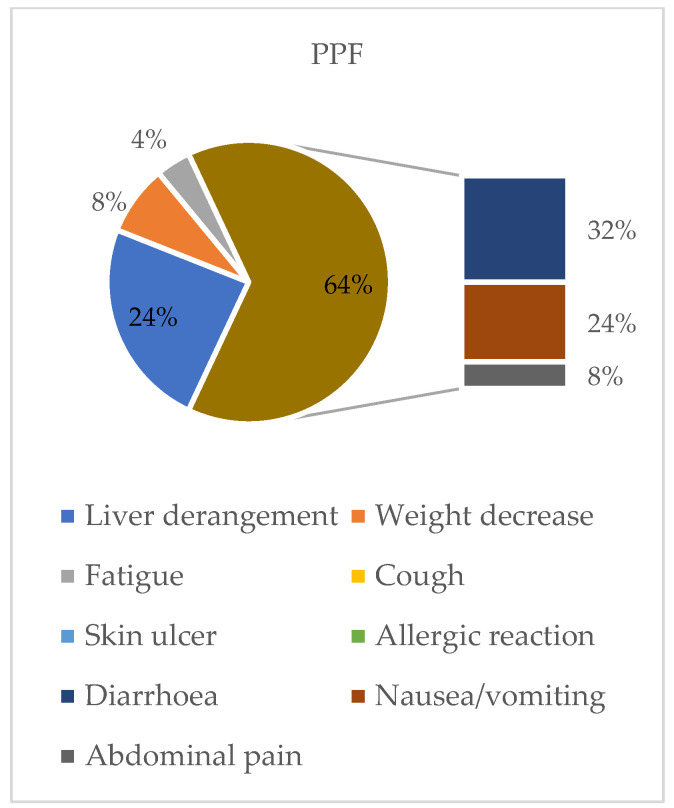
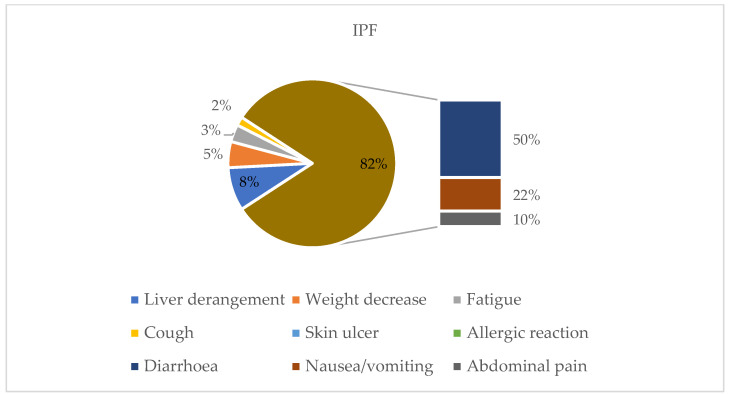
Background/Objectives: Idiopathic pulmonary fibrosis (IPF) is the most common form of pulmonary fibrosis (PF) and serves as a key reference for disease severity. Progressive pulmonary fibrosis (PPF), a distinct yet heterogeneous entity arising from various interstitial lung diseases (ILDs), shares similar pathogenetic mechanisms and clinical courses driven by self-perpetuating fibrosis. Antifibrotic therapy, notably nintedanib, can slow disease progression. However, real-world data on antifibrotic therapy's impact on survival, especially in PPF, are limited. This study aims to compare IPF and PPF regarding phenotype, radiological patterns, comorbidities, prognostic factors, and response to nintedanib, focusing on identifying the patient subsets most likely to benefit. Outcomes assessed include safety, survival, and disease progression over one year, considering various prognostic factors. Methods: This retrospective observational study evaluated patients with fibrosing ILD, affected by either IPF or PPF, and treated with nintedanib. Data collected encompassed clinical, radiological, functional, and treatment-related information. Assessments included chest CT, pulmonary function tests, comorbidities, and survival analysis, utilizing standardized methods and statistical tools to interpret outcomes and tolerability. Results: The study population was composed of 97 patients: 64 were diagnosed with IPF and 33 with PPF. The analysis showed that in PPF patients, ongoing antifibrotic treatment resulted in higher survival (71.1 months vs. 27.4 months, p < 0.001), while no statistically significant differences were found in the IPF group (67.4 months vs. 52.5 months, p = 0.216). Nintedanib was generally well tolerated. Gastrointestinal side effects, predominantly diarrhea, were reported in 61% of patients with IPF and 50% of those with PPF. Dose reduction occurred in 43.75% of IPF patients and 36% of PPF patients, while treatment discontinuation was required in 21.87% of IPF and 21% of PPF patients. Conclusions: This study highlights that in PPF patients, antifibrotic therapy with nintedanib can improve survival. This statement underlines that the primary outcome of antifibrotic treatment should focus on improving patients' survival.
期刊介绍:
Journal of Clinical Medicine (ISSN 2077-0383), is an international scientific open access journal, providing a platform for advances in health care/clinical practices, the study of direct observation of patients and general medical research. This multi-disciplinary journal is aimed at a wide audience of medical researchers and healthcare professionals.
Unique features of this journal:
manuscripts regarding original research and ideas will be particularly welcomed.JCM also accepts reviews, communications, and short notes.
There is no limit to publication length: our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


